Home, Search, Index, Links, Pathology, Molecules, Syndromes,
Muscle, NMJ, Nerve, Spinal, Ataxia, Antibody & Biopsy, Patient Info
|
Home, Search, Index, Links, Pathology, Molecules, Syndromes, Muscle, NMJ, Nerve, Spinal, Ataxia, Antibody & Biopsy, Patient Info |
|
Comparative features General features Acquired Chondrodystrophic: Perlecan; 1p34 Congenita Acetazolamide-responsive: Na+ Becker's (Recessive): Cl- Thomsen's (Dominant): Cl- Dystrophy Type I (DM1): DMPK; 19q13 Type II (DM2; PROMM): ZNF9; 3q21 Type III (DM3): VCP Fluctuans: Na+ Levior: Cl- Paramyotonia: Na+; Cl- Permanens: Na+ Potassium activated myotonia: Na Proximal Myopathy (PROMM): ZNF9; 3q21 |

|

|
Proximal Myotonic Myopathy (PROMM; DM2)
●
CCHC-type Zinc finger Nucleic acid-Binding Protein (CNBP; Zinc finger protein 9 (ZNF9))
|
|
|
Andersen: KCNJ2; 17q24 K+ channel: KCNE3; 11q13 Na+ channel: SCN4A; 17q23 |
| Mechanisms of Episodic Weakness |
|---|
|
Extracellular K+ increase:
2° to K+ intake, or Rest after exercise ↓ Membrane depolarization: Mild ↓ Na+ channels open: Abnormal Na+ channels to non-inactivating mode ↓ ↑ Persistent inward Na+ current ↑ ↓ ↑ í Extracellular K+ increase Sustained depolarization of muscle membrane î ↑ ↓ K+ efflux Inactivation of normal Na+ channels ↓ Loss of electrical excitability of muscle membrane ↓ Weakness |
|
Exercise Potassium loading Cold environment Pregnancy |
Glucocorticoids Stress Ethanol Fasting |
|
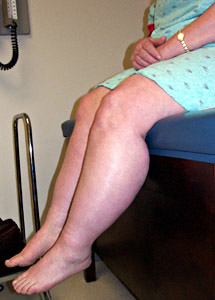
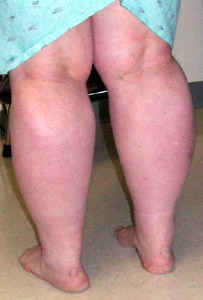
Acquired Hereditary Dominant RMD1: 1q41 RMD2: Caveolin-3; 3p25 Lipodystrophy (CGL4): Cavin; 17q21 Recessive Omani RMR: Caveolin-3; 3p25 |
 Rippling Muscles |
 From: Ho et al. BMC Neurology |
|
Familial Dwarfism + Spasms Schwartz-Jampel Sporadic Satoyoshi |
|
Normalize metabolic abnormalities Quinine sulfate: 260 mg qhs or bid Carbamazepine: 200 mg bid or tid Phenytoin: 300 mg qd Tocainide: 200 to 400 mg bid Verapamil: 120 mg qd |
Amitriptyline:
25 to 100 mg qhs Vitamin E: 400 IU qd Riboflavin: 100 mg qd Diphenhydramine: 50 mg qd Calcium: 0.5 to 1 g elemental Ca++ qd |
|
Clinical features Clostridium tetani Diagnosis Epidemiology Mechanism of action Prophylaxis Protein structure Treatment History Case description  C Bell 1865: From NLM |
 Putnam |
|
|
|
Associated disorders Clinical Differential diagnosis EMG Laboratory |
|
Antibodies Associated disorders Clinical features Laboratory Treatment Variants |
Strychnine 11
|
|
|
Muscle fibers Axon firing patterns Complex repetitive discharges Cramp Endplate Spikes Fasciculations Fibrillations Insertion activity Miniature endplate potentials (MEPPs) Positive Sharp Waves Also see: Spontaneous activity |
Neural Origin Amyotrophic Lateral Sclerosis Black widow spider Blepharospasm Complex repetitive discharges Cramp Dystonia variants Fasciculations Geniospasm Isaac's Syndrome Motor neuropathies & neuron disorders Motor units Muscle fiber activity Myokymia Neuromyotonia Polyneuropathy Startle syndrome (Hyperekplexia) Stiff-person Syndrome Tetanus Tetany Tremor Whipple's |
Tetany with carpopedal spasm
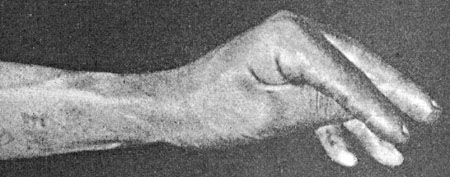 Barker |

|
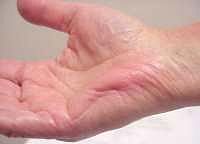 From: M. Al-Lozi |
|
Movie From: R Bucelli |
| Palmaris brevis spasm |
|
Familial cramp syndrome Familial dwarfism with muscle spasms Muscle cramp & neuropathy syndrome HANAC Cramps & myalgias |
|
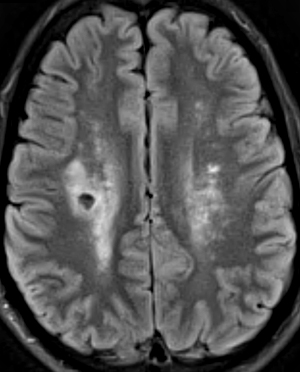
Hereditary Angiopathy, Nephropathy, Aneurysms & Muscle Cramps (HANAC)
● Collagen Type IV α1 (COL4A1)
|
|