|
Home, Search, Index, Links, Pathology, Molecules, Syndromes, Muscle, NMJ, Nerve, Spinal, Ataxia, Antibody & Biopsy, Patient Info |
AUTONOMIC DISORDERS


AUTONOMIC DISEASE SYNDROMES
- Systemic diseases
- Amyloidosis
- Diabetes Mellitus
- Porphyria
- Infections or related toxins
- Chronic renal or hepatic disease: Usually subclinical
- Immune disorders
- Acute Dysautonomia
- Guillain-Barré Syndrome
- Paraneoplastic: Especially Small cell lung cancer
- Sensory neuronopathy: anti-Hu
- Dysautonomia: IgG vs α3 subunit of acetylcholine receptor
- Enteric neuropathy: Hu; CV-2; DPPX
- LEMS
- Sjögren's syndrome
- Neurofascin-155 IgG4 antibodies: Nodopathies
- ? Collagen Vascular Disorders
- System Degenerations
- Idiopathic Orthostatic Hypotension
- Shy-Drager (Multiple systems atrophy)
- Parkinsonism: DOPA responsive
- Mitochondrial - MNGIE
(Myopathy and external ophthalmoplegia; Neuropathy; Gastro-Intestinal; Encephalopathy) - Neuronal intranuclear inclusion disease (NIID)
- Hereditary autonomic disorders
- Biemond congenital anaesthesia

- Pain (submandibular, ocular & rectal) with flushing
- Congenital dysautonomias, Other
1
- Reduced sweating
- Syncope
- Vomiting
- Reduced salivation
- Feeding difficulties during infancy
- Growth retardation
- Reduced tears
- Abnormal temperature control
- Biemond congenital anaesthesia
- Drugs & Toxins
- Polyneuropathy
- Drugs: Alcohol;
Amiodarone;
cis-Platinum;
Cyclosporine A;
Perhexiline;
Taxol; Vacor; Vincristine - Heavy metals: Arsenic; Mercury; Thallium
- Toxins: Acrylamide; Hexacarbons
- Deficiency: Vitamin B12 (may present with postural hypotension)
- Drugs: Alcohol;
Amiodarone;
cis-Platinum;
Cyclosporine A;
Perhexiline;
- Cellular dysfunction: Pharmacologic
- Reduce sympathetic activity
- Sympathetic neuron: Guanethidine; Bethanidine
- α-adrenoceptor blockade: Phenoxybenzamine; Prazosin
- β-adrenoceptor blockade: Propranolol; Timolol
- Increase sympathetic activity
- Release noradrenaline: Tyramine
- Monoamine oxidase inhibitors
- β-adrenoceptor stimulants: Isoprenaline
- Decrease parasympathetic activity
- Botulinum toxin
- Anticholinergic: Atropine; Probanthine
- Anti-arrhythmics: Disopyramide
- Increase parasympathetic activity
- Cholinomimetics
- Carbachol; Bethanicol; Pilocarpine
- Mushrooms: Muscimol & Muscarine
- Carbachol; Bethanicol; Pilocarpine
- Anticholinesterases
- Reversible: Pyridostigmine; Neostigmine
- Irreversible: Organophosphorus inhibitors
- Cholinomimetics
- Autonomic overactivity
- Reduce sympathetic activity
- Polyneuropathy
- Primary Dysautonomia: Often monophasic + remission
- Acute
- Pandysautonomia
- Pure cholinergic dysfunction
- Dysautonomia but no postural hypotension
- Postural orthostatic tachycardia syndrome (POTS)
- Gastrointestinal motility disorders
- Chronic autonomic failure
- Congenital: Nerve growth factor deficiency
- Acute
- Localized autonomic dysfunction
- Horner's syndrome: Ptosis; Miosis; Anhidrosis
- Reflex sympathetic dystrophy
- Crocodile tears (Bogorad's syndrome)
- Congenital respiratory failure
- Ross syndrome: Segmental anhidrosis; Hyporeflexia; Tonic pupils
AUTONOMIC SYNDROMES: System disorders
|
Acute Cardiac Gastrointestinal Hypotension Lacrimation Ocular Pandysautonomia Respiratory Sexual Sweating Urinary |
|
Pandysautonomia: Autoimmune; Acute, Sub-Acute or Chronic 16
- Onset
- Age: Adult or Child
- Signs
- Most common: Postural hypotension (78%); GI dysfunction (70%); Anhidrosis
- Other: Urinary; Impotence
- Females > Males 2:1
- Progression over 1 to 8 weeks
- "Viral prodrome" in 59%: Especially URI or Flu-like syndrome
- Previously healthy person
- Anatomical features
- Distribution of autonomic dysfunction: Diffuse; Truncal; Scalp
- Autonomic systems involved: Sympathetic & Parasympathetic in 80%
- Clinical features
- Orthostatic hypotension: Without compensatory tachycardia
- Gastrointestinal dysmotility (80%)
- Anhidrosis (60% to 74%)
- Distribution: Diffuse or Distal predominant
- Post-ganglionic
- Heat intolerance
- More with higher α3-AChR antibody titers
- Dry eyes & mouth (50%)
- Pupillary response reduced (30%)
- GU: Urinary or Erectile dysfunction (30%)
- Sensory (25%)
- Reduced Small fiber modalities
- Paresthesias in extremities
- Cough
- Cognitive impairment
- Course: Chronic persistent deficits or Monophasic
- Laboratory
- CSF: Elevated protein in 60%; No cells
- Antibody: IgG vs α3
subunit of acetylcholine receptor
4 (30%)
- Epidemiology
- Frequency: 50% of acute onset pure autonomic syndromes
- Systems involved: Sympathetic & Parasympathetic
- Clinical
- Initial features
- Orthostatic: Blood pressure or pulse changes
- GI: Motility disorders
- Other features
- Sudomotor: Postganglionic anhidrosis
- Dry eyes & Mouth
- Pupillary disorders
- Urinary bladder dysfunction
- Course: Monophasic
- Initial features
- Antibody titers
- Higher (≥ 1 nmol/L) correlate with
- Autonomic features: Presence & More severe
- Neoplasm
- Low titers (0.03-0.09 nmol/L)
- Autonomic syndromes: Limited
- Not specific for autonomic disorders
- Sero-negative
- Persistent: Orthostatic hypotension; GI dysfunction
- Higher (≥ 1 nmol/L) correlate with
- Neoplasms (30%): Breast; Lymphoma
- Other disorders with anti-α3-AChR antibody: Especially intermediate titers
- Isaacs syndrome: 50%
- Lambert-Eaton syndrome: 10%
- Myasthenia gravis: Some patients
- Polyneuropathy
- Encephalopathy
- Lung cancer: 8%
- Common associated antibodies
- GAD-65
- Muscle AChR
- Voltage-gated cation channel
- N-type Ca++
- P/Q type Ca++
- K+-associated
- Striational
- Hu
- CRMP-5
- Also see: AChR disorders
- Epidemiology
- Nerve pathology: Occasional perivascular inflammation
- Disease associations
- Differential diagnosis
- Recovery: Variable degrees
- Partial: Most common outcome
- Slow over months to years;
- Correlates with: Reduction in Antibody titer
- Decreased Blocking of AChR function
- None: Some patients
- Complete recovery: Occasional
- Partial: Most common outcome
- Treatment
- For any associated neoplasm
- Pharmacologic or Other: Anecdotal evidence
- IV Ig; Especially in 1st 8 weeks of disease or progressing disability
- Corticosteroids
- Plasma Exchange
- Other immumodulating medications
- Animal model: Rabbits immunized vs recombinant α3 nAChR subunits
15
- Clinical features: GI hypomotility; Dilated pupils with impaired light response; Distended bladders
- Severity parallels serum levels of ganglionic nAChR autoantibody
- Post-synaptic defect
- Failure of neurotransmission through abdominal sympathetic ganglia
- Retention of neuronal viability
Dysautonomia + CNS autoimmune syndrome 33
- Antibody & Target: IgG vs DPPX (DPP6)
- Tissue staining: Synaptic
- Antibody location: Serum ± CSF
- DPPX protein
- Dipeptidyl-peptidase-like protein-6 (DPPX)
- Regulatory subunit of neuronal Kv4.2 (KCND2) potassium channels
- Clinical
- Onset
- Age: Mean 53 years
- Course: Insidious (75%), Sub-acute (25%)
- CNS (100%)
- Cortical function: Amnesia, Delirium, Psychosis, Depression
- Hyperexcitability: Myoclonus, Exaggerated startle, Rigidity, Hyperreflexia, Seizures
- Brainstem: Eye movement disturbances, Ataxia, Dysphagia, Dysarthria, Respiratory failure
- Sleep disorders: Insomnia, Periodic limb movements, Sleep apnea, Hypersomnia
- Autonomic (50%)
- GI: Diarrhea, Gastroparesis, Constipation
- Bladder
- Cardiac conduction disorder
- Occasional: Diaphoresis; Thermoregulation
- Weight loss (50%)
- Sensory disorders (10%): Pruritis; Allodynia; Paresthesias
- Lymphoma & B-cell neoplasm (10%)
- Treatment: Immunotherapy effective in 60%
- Onset
- Laboratory
- Brain MRI: Normal or Non-specific WM changes
- EEG: May be normal
- CSF: Cells 40%; Protein may be high
Gastrointestinal syndromes
|
- Hirschsprung's congenital megacolon: General
- Epidemiology
- Incidence: 1 in 5,000 newborns
- Male predominance: 4 to 1
- Sibling recurrence: Usually 3% to 4%; 200x higher than general population
- General features: Pathophysiology
- Absent intrinsic ganglion cells in the submucosal & myenteric plexuses
- Over variable lengths of the distal gut
- Failure in time-specific migration of neural-crest derived ganglion cells into GI tract
- Gene pathways involved
- RET receptor tyrosine kinase: RET receptor; GDNF (RET receptor ligand)
- Endothelin type B receptor: EDNR; Endothelin-3 (EDNR ligand)
- SOX-10 mediated transcription
- Absent intrinsic ganglion cells in the submucosal & myenteric plexuses
- Genetics
- Susceptibility loci (HSCR)
- Inheritance patterns: Non-syndromic disease may be oligogenic
- Aganglionosis only in sigmoid colon: Often sporadic; 60% to 85% of cases
- Aganglionosis in long length of intestine: More likely familial occurrence
- Clinical features
- General: Isolated HSCR 80%; Syndromic HSCR 20%
- Onset: Congenital
- Constipation
- Vomiting
- Abdominal distention
- Intestinal obstruction
- Associated congenital malformations (30%)
- Cranial pigment formation
- Sensory components of acoustic pathway
- Pathology
- Absent enteric parasympathetic ganglia along a variable length of intestine
- Incomplete migration of neurenteric ganglion cells from neural crest to gut
- Acetylcholinesterase: Increased in submucosal & myenteric plexus of affected segment
- Narrowed distal segment of bowel
- Diagnosis: Absent or reduced ganglion cells in affected segment in rectal biopsy
- Absent enteric parasympathetic ganglia along a variable length of intestine
- Epidemiology
- Hirschsprung disease, susceptibility to, 2 (HSCR2)
● Endothelin receptor type B (EDNRB; HSCR2);
Chromosome 13q22.3; Recessive
- Genetics
- Penetrance: 30% to 85%
- Allelic disorders
- Waardenburg-Shah syndrome 4A (WS4A)
- ABCD syndrome
- Waardenburg-Shah syndrome 4A (WS4A)
- EDNRB protein
- Role in: Schwann cell differentiation
- Pain relations
- Stimulus: Actions on endothelin-A receptors of local nociceptors
- Analgesia: Endothelin-B receptors
- Clinical
- GI (Hirschprung's): Short segment bowel disease in 95%
- Deafness
- Pigmentary anomalies
- Genetics
- Hirschsprung disease, susceptibility to, 3 (HSCR3)
● Glial cell line-derived neurotrophic factor (GDNF);
Chromosome 5p13.2; Dominant or Recessive
- Genetics
- ? Some cases have digenic inheritance with RET
- ? Penetrance
- Allelic disorders
- Central hypoventilation syndrome, Dominant
- Pheochromocytoma, modifier of, Dominant
- Central hypoventilation syndrome, Dominant
- Also see: Autonomic sleep ventilation failure
- Genetics
- Congenital central hypoventilation with Hirschsprung disease (HSCR4)
● Endothelin-3 (EDN3);
Chromosome 20q13.32; Recessive or Dominant with reduced penetrance
- Genetics: Allelic disorders
- Waardenburg-Shah syndrome 4B (WS4B)
- Hirschsprung disease, susceptibility to, 4
- Waardenburg-Shah syndrome 4B (WS4B)
- Clinical
- Autonomic sleep ventilation failure
- GI: Megacolon (Often whole length aganglionosis); Esophageal motility reduced
- Face: Antimongoloid eye slant; Triangular mouth
- Low-set ears; Small nose
- Heart rate control: Reduced
- Differential diagnosis
- Depression of ventilatory response to hypoxia and hypercapnia
- Familial lethal sleep apnea
- Depression of ventilatory response to hypoxia and hypercapnia
- Genetics: Allelic disorders
- Hirschsprung disease, susceptibility
● Neurturin
Chromosome 19p13.3; Sporadic
- ? Some cases have digenic inheritance with RET
- Hirschsprung disease, cardiac defects, and autonomic dysfunction
● Endothelin-converting enzyme (ECE1);
Chromosome 1p36.1; Dominant or Recessive
- Genetics
- Mutation: Arg742Cys
- Allelic with: Essential hypertension, susceptibility
- ECE1 protein
- Involved in proteolytic processing of endothelin-1
- Clinical
- Hirschsprung disease: Skip-lesions
- Cardiac defects
- Craniofacial abnormalities + Other dysmorphic features
- Autonomic dysfunction: episodic
- Agitation
- Tachycardia
- Fever
- Genetics
- Hirschsprung disease, susceptibility to, 10 (HSCR10)
● SOX10;
Chromosome 22q13.1; Dominant or Sporadic (Recessive)
- Genetics
- Mutations: Upstream regulatory region; 38-kB Del, 38412781G-C, 38412215G-A
- Allelic with
- Waardenburg syndrome, type IIE (WS2E)
- Waardenburg syndrome, type 4C (WS4C)
- Yemenite deaf-blind hypopigmentation syndrome
- Waardenburg-Shah syndrome, neurologic variant
- Waardenburg syndrome, type IIE (WS2E)
- Clinical
- Hypopigmentation
- Deafness
- Enteric aganglionosis
- Laboratory
- Hypomyelination in CNS & PNS
- Genetics
- Visceral Neuropathy 2 (VSCN2) (Hirschsprung disease (HSCR))
47
● ERB-B2 Receptor tyrosine kinase 2 (ERBB2; Herstatin; Neu; Her2);
Chromosome 17q12; Recessive
- Epidemiology: 1 family, 2 patients
- Genetics
- Mutation: Missense; c.2129C>T, Ala710Val; Homozygous
- Allelic disorders: Somatic mutations in neoplasms
- ERBB2 protein
- Present in enteric neurons
- Locations: Nucleus & Endosome
- IL6 signaling through MAP kinase pathway
- ErbB receptor tyrosine kinase component
- Over-expression in neoplastic transfoprmation of prostate cancer
- Activated in nerve distal to transection
- Schwann cell precursors
- Breast cancer oncogene
- M. leprae binds to extracellular domain of ERBB2
- Clinical
- Arthrogryposis: Club feet
- Polyneuropathy
- Hearing loss
- Ptosis
- Hirschsprung disease: Short segment
- Laboratory
- GI: Hypoganglionosis
- Mowat-Wilson syndrome
● SMADIP1 (Zinc finger homeobox 1B; ZFHX1B; ZEB2)
Chromosome 2q22.3; Dominant > Recessive
- Genetic: Mutations
- Frameshift or Nonsense
- Deletion syndrome: Occasional patient
- SMADIP1 protein (ZFHX1B; ZEB2)
- 2-handed zinc finger/homeodomain proteins (Zinc finger homeo box 1B)
- SMADIP1 interacts with receptor-mediated, activated full-length SMADs
- Transcriptional repressor
- Required for: Schwann cell differentiation & myelination
- Clinical
- May appear in isolation (Non-syndromic) or with other systemic
- Neonatal: Hypotonia; Hirschsprung megacolon
- Facial phenotype: Broad nasal bridge (Hypertelorism); Open mouth; Triangular jaw
- EENT: Large, deep-set eyes (Blue); Large uplifted earlobes
- CNS
- Mental retardation: Minimal speech; Delayed motor
- Epilepsy
- Learning problems
- CNS Pathology: Microcephaly; Corpus callosum agenesis
- Ocular: Iris coloboma; Ptosis; Bicolored irides
- Heart disease: Congenital
- Similar recessively inherited syndrome: Goldberg-Shprintzen
- Genetic: Mutations
- Goldberg-Shprintzen Megacolon syndrome (GOSHS)
with Polymicrogyria
22
● Kinesin family binding protein (KIFBP; KIF1BP; KIAA1279) ; Chromosome 10q22.1; Recessive
; Chromosome 10q22.1; Recessive
- Epidemiology: > 45 patients
- Genetics
- Mutations: Loss of function; Stop, Deletion& Missense; Arg90X, Glu84X, Ser200X
- KIFBP protein
49
- Interacts with: Microtubules; Kinesins, SCG10
- Tetratrico peptide repeat (TPR) family
- Ubiquitous expression
- Functions: Regulates
- Axon microtubules: Organization & Dynamics
- Kinesin attachment
- Mitochondria: Biogenesis & Distribution
- Mitosis: KIF15 & KIF18 interactions
- Cytokinesis: Citron Kinase (CIT)
interactor
- Other TPR family disorders
- Leber congenital amaurosis: AIPL1
- Charcot-Marie-Tooth 4C: KIAA1985
- Clinical
- CNS
- Microcephaly
- Mental retardation
- GI: Hirschsprung disease (70%)
- Face Dysmorphic: Broad nasal bridge, Hypertelorism, Synophrys, Large ears, Long nose
- Peripheral neuropathy: Axon loss
- Short stature
- Other inconstant features
- Eye: Megalocornea, Iris coloboma, Corneal ulcers; Ptosis, Arched eyebrows, Dense eyelashes
- Urogenital anomalies
- Similar Dominantly inherited syndrome: Mowat-Wilson
- CNS
- Laboratory
- Brain pathology: Micropolygyria; Corpus callosum hypoplasia
- Hirschsprung susceptibility (HSCR1)
● RET oncogene (RET);
Chromosome 10q11.21; Dominant or Sporadic
- Frequency: 50% of familial cases; 15% to 35% of sporadic cases
- Genetics
- Gene mutations
- Multigenerational families: Mutations occur throughout gene
- Sporadic cases: Mutations in region encoding extracellular domain
- Genetic mechanism
10
- Loss of function
- Association with other gene mutations
- More severe, longer segment disease with additional Neurturin
mutation
- Other associated genetic loci (susceptibility)
- Locations
- HSCR6
; Chromosome 3p21
- HSCR7
; Chromosome 19q12
- Hirschsprung genetic modifier 8 (HSCR8)
: Chromosome 16q23
- Hirschsprung genetic modifier 9 (HSCR9)
:
Chromosome 4q31.3-q32.3
- HSCR6
- Transmission: Maternal > Paternal
- Magnitude of effects: Multiplicative among loci
- Locations
- More severe, longer segment disease with additional Neurturin
- Penetrance: 50% (Females) to 72% (Males)
- Allelic disorders
- Central hypoventilation syndrome, congenital
- Medullary thyroid carcinoma
- Multiple endocrine neoplasia IIA
- Multiple endocrine neoplasia IIB
- Pheochromocytoma
- Renal agenesis
- Central hypoventilation syndrome, congenital
- Gene mutations
- Clinical
- Often non-syndromic Hirschsprung's congenital megacolon
- Short segment bowel disease in 25%
- Autonomic sleep ventilation failure
- Hirschsprung syndromes: Other
- Down syndrome-associated
- Polydactyly, Renal agenesis, Deafness
- Hypoplastic nails or finger deformities
- Multiple endocrine neoplasia 2A
;
2B
&
Medullary thyroid carcinoma
- Heart defects, Laryngeal anomalies, Preaxial polydactyly
- Type D brachydactyly
- Pitt-Hopkins
- Achalasia
- Definition
- Absence of peristalsis in esophagus
- Failure of relaxation of lower esophageal sphincter
- Etiology
- Usually 1° disorder
- Achalasia-Addisonianism-Alacrimia syndrome: AAAS
- Achalasia-Alacrimia-Intellectual Dysfunction: GMPPA
- Achalasia-Alacrimia-Peripheral Neuropathy: NDC1
- Autoimmune polyendocrinopathy-Candidiasis-Ectodermal dystrophy (APECED): AIRE
- Achalsia, Early onset + Moya Moya: GUCY1A3
- Achalasia-Microcephaly
- Familial achalasia, esophageal, Recessive
- Achalsia-Progeria/Lipodystrophy
 : BUD13
: BUD13
- Occasionally 2° to Chagas
- Usually 1° disorder
- Pathology
- Loss of ganglion cells & myenteric nerves
- Reduced numbers of vasoactive intestinal peptide-containing myenteric plexus neurons
- Definition
- Multiple Endocrine Neoplasia 2b (or 3)
● Rearranged during transfection protooncogene (RET);
Chromosome 10q11.21; Dominant
- Genetics
- Common mutation: M918T
- Allelic disorders
- Central hypoventilation syndrome, congenital
- Medullary thyroid carcinoma
- Multiple endocrine neoplasia IIA
- Pheochromocytoma
- Renal agenesis
- Hirschsprung disease, susceptibility
- Central hypoventilation syndrome, congenital
- Clinical
- Morphology: "Marfanlike" body build (Long arms), Full & fleshy lips
- GI dysmotility: Esophagus, Megacolon
- Leg atrophy: Distal
- Neuropathy
- Autonomic; Some sensory & motor
- Axon loss
- Neoplasm associations
- Medullary Thyroid
- Pheochromocytoma
- Ganglioneuromas: Tongue & Lips; GI tract; Corneal
- Genetics
- Visceral myopathy with External Ophthalmoplegia, Familial
● Recessive- Epidemiology: 3 German families
- Clinical
- Eyes: Ptosis & ophthalmoplegia
- GI: Intestinal pseudoobstruction; Ascites
- Progression: Death often < 30 years
- Pathology
- GI: Loss of smooth muscle in stomach & intestines
- Peripheral neuropathy
- Visceral Myopathy, Familial 1 (VSCM1)
● Actin, γ-2, Smooth muscle, enteric (ACTG2; ACTA3); Chromosome 2p13.1; Dominant, Recessive or Sporadic
- Nosology
- Megacystis-microcolon-intestinal hypoperistalsis syndrome (MMIHS)
- Chronic Intestinal Pseudo-Obstruction (CIPO): Congenital or Late onset
- Epidemiology
- > 25 families
- 45% to 50% of CIPO
- Genetics
- Mutations: Missense; Often de novo
- Null alleles: Recessive
- Severe: Arg178
- Allelic disorders
- Megacystis-Microcolon-Intestinal Hypoperistalsis Syndrome-5 (MMIHS5)
- Possible variant: African Degenerative Visceral Leiomyopathy (ADL)
- Mutations in ACTG2 & RET identified
- Megacystis-Microcolon-Intestinal Hypoperistalsis Syndrome-5 (MMIHS5)
- ACTG2 protein
- Actin
- Higher transcription levels in tissues with smooth muscle
- Enteric muscle contraction
- Mutations: Interfere with polymerization of ACTG2 into thin filaments
- Clinical
- Onset age: Prenatal or Infancy
- GI
- Constipation
- Intestinal dysmotility
- Megacolon
- Intestinal malrotation
- Urinary
- Megacystis
- Hydronephrosis
- Course: Persistent into adulthood
- Treatment: Surgical; Transplantation
- Laboratory
- Jejunum: Outer muscular layer thin
- GI ganglion cells: Present
- Nosology
- CIPO type: Chronic Atrial & Intestinal Dysrhythmia
● Shugoshin-like 1 (SGOL1):
Chromosome 3p24.3; Recessive
- Epidemiology: 17 patients, 14 families
- Genetics
- Mutation: Missense; Lys23Glu
- SGOL1 protein
- Location: Nucleus
- Mitotic progression
- Chromosome cohesion & segregation during mitosis
- Prevents premature dissociation of cohesin complex from centromeres after prophase
- Clinical
- Onset age: Usually 1st or 2nd decade
- Cardiac: Dysregulation of cardiac sinus node; Sinus bradycardia
- GI: Chronic intestinal pseudoobstruction (CIPO)
- Laboratory
- GI pathology: Ganglia mislocalized & hypoplastic
- Intestinal pseudoobstruction, neuronal, chronic idiopathic, X-linked (CIPO; CIIPX)
● Filamin A (FLNA):
Chromosome Xq28; Recessive
- Epidemiology: > 30 patients
- Genetics
- Mutations: Duplications of Xq28 including FLNA; Stop; Missense
- Allelic disorders
- ?FG syndrome 2
- Cardiac valvular dysplasia, X-linked
- Congenital short bowel syndrome, Recessive
- Frontometaphyseal dysplasia 1, Recessive
- Heterotopia, periventricular, Dominant
- Intestinal pseudoobstruction, neuronal, Recessive
- Melnick-Needles syndrome, Dominant
- Otopalatodigital syndrome, type I, Dominant
- Otopalatodigital syndrome, type II, Dominant
- Terminal osseous dysplasia, Dominant
- ?FG syndrome 2
- FLNA protein
- Down-regulates: Androgen receptor function
- Location: Cytoplasmic or Nuclear
- Transcription regulation
- Mediates crosslinking actin filaments
- Connects cytoskeleton to membrane glycoproteins
- Apoptosis-related: Neuronal; Spinal cord injury
- Regulates proliferation dynamics of neural progenitor cells: Modulates Cdk1 activity
- PI3K/AKT signalling pathway
- Interacts with: FLNC; FILIP1
- Clinical
- Onset: Early childhood
- Bowel malrotation
- CNS
- Developmental delay
- Spasticity
- Visceral neuropathy, Familial (VSCN1)
● ERBB3; Recessive
- Visceral neuropathy 3, Familial (VSCN3)
● Dominant- Epidemiology: 4 families
- Clinical
- Onset age: 1st to 7th decades
- Eyes: Ptosis; Pupils unresponsive
- GI
- Intestinal pseudo-obstruction
- Esophageal achalasia
- Abdominal distention
- Polyneuropathy
- Sensory loss
- NCV: Axon loss
- Pathology: Myenteric plexus
- Neurons: Abnormal morphology; Increased number
- Nerve trunks & Myenteric ganglia: Hyperplastic
- Cartilage-hair hypoplasia (CHH)
● Mitochondrial RNA-processing endoribonuclease, RNA component (RMRP);
Chromosome 9p13.3; Recessive
- Epidemiology: Old Order Amish (US); Finland
- Genetics
- Mutations
- Insertions & Deletions; In & out of frame
- Finnish & Old Order Amish: A70G
- Allelic with
- Metaphyseal dysplasia without hypotrichosis
- Anauxetic dysplasia
- Metaphyseal dysplasia without hypotrichosis
- Mutations
- RMRP
- RMRP gene: Untranslated, encodes RNA, not protein
- Ribonucleoprotein: Contains protein & RNA components
- RNA encoded by single-copy gene in nucleus: Imported into mitochondria
- Enzyme activity
- Endoribonuclease
- Cleaves mitochondrial RNA complementary to light chain of displacement loop
- Clinical
- Skeletal
- Short stature: Disproportionate
- Joint hyperextensibility: Hands, wrist, & feet
- Metaphyseal dysplasia: May be only manifestation
- Hair: Hypoplastic; Fine, sparse & light-colored
- Autonomic
- Intestine: Neuronal dysplasia; Megacolon; Malabsorption
- Malignancy: Increased risk of lymphoma & skin neoplasm
- Skeletal
- Laboratory
- Defective immunity: Susceptibility to chickenpox; Lymphopenia; Neutropenia
- Anemia: Hypoplastic macrocytic
- Acquired GI disorders
- Chagas' disease (Trypanosomiasis cruzii)
- Gustatory sweating (Frey's syndrome)
- Paraneoplastic
- Post-surgical
- Regional sympathectomy
- Dumping syndrome: Post-vagotomy & gastric drainage
- Heart transplantation
Heart rate: Parasympathetic control
- Tachycardia
- Fixed:
Diabetes mellitus;
Amyloidosis
- Cardiac parasympathetic failure
- Guillain-Barré
- Flavin-containing monooxygenase 3
- Fish odor syndrome
- Tachycardia & hypertension after eating cheese containing tyramine
- Trimethylaminuria produces fish odor
- Fish odor syndrome
- Postural Orthostatic Tachycardia Syndrome (POTS)
- Cardiomyopathy: Dystrophinopathy
- Normal
- Inspiration
- Magnitude of changes with inspiration & expiration decreases with age
- Fixed:
Diabetes mellitus;
Amyloidosis
- Bradycardia
- Resting: Dopamine β-hydroxylase deficiency
- Parasympathetic sparing
- Response to stimuli
- Carotid sinus hypersensitivity
- Swallow & vasovagal syncope
- Expiration (Vagal; Cholinregic)
- Cardiomyopathy: Emery-Dreifuss
- Resting: Dopamine β-hydroxylase deficiency
Postural hypotension (Blood pressure: Sympathetic, noradrenergic control)
- Causes
- 1° Autonomic dysfunction
- Hereditary
- Hereditary Orthostatic Hypotension
- Amyloidosis
- Epithelial sodium channel genes: Pseudohypoaldosteronism
- Mitochondrial disorder: Maternal inheritance
- Postural Orthostatic Tachycardia Syndrome (POTS): SLC6A2
- Spastic-Ataxia with Lipodystrophy: CAV1
- Mast cells: TPSAB1
- Adult-onset Leukodystrophy: LMNB1
- Sporadic
- Hereditary
- Hypotension
- Corticosteroid-binding globulin deficiency
: Fatigue; Hypotension
- Corticosteroid-binding globulin deficiency
- Low intravascular volume
- Vasodilation
- Reduced cardiac output
- Drugs: Amphetamines; Vinca alkaloids
- 1° Autonomic dysfunction
- Exacerbation
- Rapid positional change
- Morning
- Large meals
- Prolonged recumbency
- Warm environment
- Increased intrathoracic pressure: cough; micturition; defecation; exertion
- Vasoactive drugs
- Treatment
39
- Avoid exacerbating factors
- Avoid: Alcohol; Sugary beverages
- Correct anemia
- Physical: Compression Stockings; Abdominal binder
- Reduce salt loss: Mineralocorticoids
- Fludrocortisone 0.1 mg qd to qid
- Vasoconstriction
- Resistance vessels
- Midodrine (α-adrenergic agonist) 10 mg bid or tid
- Phenylephrine
- Phenylpropanolamine 25 mg tid
- Capacitance vessels: Dihydroergotamine
- Indirect: Ephedrine; MAO inhibitors; Yohimbine
- Resistance vessels
- Prevent vasodilation
- Prostaglandin synthetase inhibitors: Indomethacin; Flurbiprofen
- Dopamine receptor blockade: Metoclopramide; Domperidone
- β-2 adrenoceptor blockade: Propranolol
- Prevent postprandial hypotension
- Avoid gastric filling: Smaller more frequent meals
- Adenosine receptor blockade: Caffeine 250 mg
- Strong coffee or tea before arising from bed & with meals
- Water drinking
9
- Acts rapidly (Minutes)
- Before meal: 120 to 480 ml over 5 minutes
- Total daily intake: 2 to 3 liters
- Mediated through sympathetic activation: Increased plasma norepinephrine
- Peptide release inhibitors: Somatostatin analogue; Octreotide
- Other medications: Ibuprofen 400 mg; Phenylpropanolamine 25 mg; Ergotamine Intranasal: 1 or 2 puffs
- Increase cardiac output: Pindolol; Xamoterol
- Increase red cell mass: Erythropoietin
- Reduce nocturnal polyuria: Desmopressin
- Reduce fall in diastolic pressure: Pyridostigmine (60 mg) 24
- Avoid supine hypertension:
- Treatment: ACE inhibitor with maximal dose at bedtime
- Pyridostigmine bromide
- Enalapril
- Reduce excessive fludrocortisone
- No vasoconstrictors after 6 pm
- Sleep with head of bed elevated
- Treatment: ACE inhibitor with maximal dose at bedtime
- Avoid exacerbating factors
Other blood pressure disorders
- Epithelial sodium channel genes
SCNN1A
;
SCNN1B
;
SCN1G
- Hypotension: Pseudohypoaldosteronism, Type 1
- Hypertension: Liddle syndrome
Sweating 40
|
General Hyperhidrosis Acquired Hereditary CNS Anhidrosis Differential diagnosis Drugs Hereditary |
- General
- Mechanism of evaporative heat loss
- From: Eccrine glands
- Control
- Afferent
- Peripheral: Thermoreceptors in skin & viscera
- CNS: Spinothalamic tract to reticular formation; Medial preoptic area of the hypothalamus
- Efferent
- CNS: Rostral ventromedial medulla; Spinal cord intermediolateral column
- Peripheral
- Autonomic type: Sympathetic
- Transmitters: Muscarinic cholinergic (ACh); Cholinergic M3-type receptors
- Anatomy: Face (T1-T4; Arms (T2-T8; Trunk (T4-T12; Legs (T10-T12)
- Pathway: White rami communicantes to paravertebral sympathetic
- Afferent
- Sweating Types
- Thermoregulation: Most eccrine sweat glands on forehead & arms
- Emotional: Palms & Soles
- Hyperhidrosis
- Hyperhidrosis Definiton: Sweating above necessary to maintain core temperature
- Mechanisms
- Aquaporin-5 (AQP5)
in sweat gland plasma membranes necessary for secretion of sweat
- Aquaporin-5 (AQP5)
- General disorders
- Essential generalized
- Compensatory for loss of sweating elsewhere
- May be initial symptom with progression to anhydrosis
- Gustatory sweating (Frey's syndrome; Baillarger’s syndrome)
50
- Pathologies
- Aberrant parasympathetic, cholinergic axons supply (facial) sweat glands & vessels
- Auriculotemporal nerve: Aberrant regeneration by parasympathetic axons
- Damaged sympathetic nerve fibers → ↑ Sweat gland sensitivity
- Sweat glands stimulated by Ach released from adjacent parasympathetic axons
- Etiologies
- Hereditary, Dominant: 1 family
- Acquired
- Parotid lesions: Especially neoplasms & parotidectomy
- Diabetes
- Age: > 45 years
- Hereditary, Dominant: 1 family
- Clinical
- Onset ages: Child & Adult
- Stimuli: Spicy foods; Independent of food type; Smell
- Sweating distribution
- Face ± Head or Upper body
- Superior cervical ganglion area
- Onset: End of, or After, meal
- Duration: 10 to 30 minutes
- Other features: Face warmth; Flushing; Pruritis
- Treatment: Botulinum toxin
- Course: Stable or Regression (Especially children)
 Lucja Frey 1918
Lucja Frey 1918 - Pathologies
- Toxic
- Other acquired
- POEMS syndrome
- Neuromyotonia
- Holmes-Adie
- Harlequin syndrome
- Stiff-person syndromes (PERM)
- Axillary sweating
- Segmental hyperhidrosis
- Causes: Syringomyelia; Vascular disorders; Neoplasms
- Possible treatments: Botulinum toxin; Mexiletine
- Hirayama
- Endocrine
- Acromegaly
- Hyperthyroid
- Menopause
- Hypoglycemia
- Diabetes
- Neoplasm
- Carcinoid
- Pheochromocytoma
- Hematologic: Lymphoma; Leukemia; Castleman
- Renal cell
- Medications
- Direct: Propranolol, Physostigmine, Pilocarpine, Bethanechol,
Tricyclic antidepressants, Serotonin reuptake inhibitors, Proton pumpo inhibitors - Drug withdrawal: Opiates; Alcohol
- Neuroleptic malignant syndrome
- Direct: Propranolol, Physostigmine, Pilocarpine, Bethanechol,
- Nocturnal
- Infection: Tuberculosis; Endocarditis
- Neoplasm: Lymphoma
- Endocrine: Diabetes mellitus; Acromegaly; Menopause
- Other: Obstructive sleep apnea; Prinzmetal angina
- Focal
- Dermatologic
- Pretibial myxedema
- POEMS
- Hereditary
- HSAN IIB
- Riley-Day syndrome (HSAN 3)
- Hereditary Sensory > Motor Neuropathy with Ulcero-mutilation
- Congenital absence of pain
- SPOAN
- HUMOP2
- Cold-Induced Sweating syndromes (CISS)
- Hyperhidrosis, Palmoplantar
● Chromosome 14q11.2-q13; Dominant- Epidemiology: Common in Japanese in Hawaii & Chinese
- Nosology: Primary focal hyperhidrosis (PFH)
- Genetics: Linkage in 3 of 11 families
- Clinical
- Onset age: Childhood
- Distribution
- Linked families: Mainly palmar
- Exacerbated by: Emotion
- Not present at night
- Primary focal hyperhidrosis
29
● Autosomal Dominant- Epidemiology: Northern Italian family
- Genetics: Not linked to 14q
- Clinical
- Onset age: 4 to 12 years
- Distribution: Palms; Axilla; Soles
- Other autonomic
- Intermittent toe blanching & coldness
- Orthostatic symptoms
- Exacerbation: Warm environment; Stress
- No sweating during sleep
- Course: Progression over decades
- Laboratory
- Sympathetic & Parasympathetic dysfunction
- Skin: Reduced innervation, proximal & distal
- Hypertrophic Osteoarthropathy, primary, autosomal recessive, 1 (PHOAR1)
● 15-Hydroxyprostaglandin dehydrogenase (HPGD; PDGH1); Chromosome 4q34.1; Recessive
- Epidemiology: Male > Famale
- Genetics: Allelic with Cranioosteoarthropathy & Digital clubbing
- Clinical
- Onset: Birth
- Skin
- Hyperhidrosis
- Oily
- Thickened
- Flushing
- Hyperkeratosis
- Head & Neck
- Face: Coarse; Ptosis; Prominent folds
- Palate: High arched
- Skeletal
- Fingers: Clubbing
- Bones: Multiple disorders
- Joints: Arthralgias; Reduced mobility
- Laboratory
- Prostaglandin E2: Increased in urine
- Mal de Meleda
● Secreted LY6/Plaur domain-containing protein (SLURP1); Chromosome 8q24.3; Recessive
- Epidemiology: High frequency in Island of Meleda, Dalmatia, Yugoslavia
- SLURP1 protein
- May interact with neuronal acetylcholine receptors (CHRNA7
)
- Secreted
- May interact with neuronal acetylcholine receptors (CHRNA7
- Clinical
- Onset age: Early infancy
- Mouth: Perioral erythema
- Hands: Brachydactyly
- Skin
- Congenital symmetrical palmoplantar keratosis
- Ichthyosis
- Hyperhidrosis
- Lichenoid plaques
- Nails: Fragile, Lusterless
- Odontoonychodermal dysplasia (OODD)
● Wingless-type mmtv integration site family, member 10A (WNT10A); Chromosome 2q35; Recessive
- Clinical
- Tongue: Smooth; Reduced papillae
- Hypodontia
- Hyperhidrosis: Palms & Soles
- Hyperkeratosis
- Nails: Dystrophic; Absent
- Hair: Absent at birth
- Clinical
- Nail disorder, nonsyndromic congenital, 5 (NDNC5)
● Autosomal Dominant- Nosology: Hereditary distal onycholysis
- Clinical
- Nails
- Growth: Decreased rate
- Scleronychia
- Detachment: Straight or concave proximal edge
- Hyperhidrosis: Palmoplantar
- Cold Sensitivity: Fingers
- Nails
- Book syndrome
● Autosomal Dominant- Premolar aplasia
- Hyperhidrosis
- Canities prematura
- Gamstorp-Wohlfart Syndrome
● Autosomal Dominant- Myokymia
- Myotonia
- Muscle wasting
- Hyperhidrosis
- CNS Related
- Shapiro syndrome: Essential Hypothermia with Hyperhidrosis
- Acquired: Post-traumatic or Post-Hemorhagic
- Fatal familial insomnia
- Parkinson disease
- Anhidrosis
- General: Due to damage to
- Preganglionic sympathetic efferent axons
- Unmyelinated, postganglionic, cholinergic axons in peripheral nerves
- Sweat glands in skin
- Differential diagnosis
Distribution Etiology of Anhidrosis Pathology Distal Polyneuropathies Postganglionic Proximal Shy-Drager Central
PreganglionicDiffuse Amyloidosis
Chronic idiopathic dysautonomia
Congenital sensory neuropathy
Cystinosis
Diabetes insipidus
Ectodermal dysplasia
Fabry's
Familial (Normal sweat glands)
Harlequin syndrome
Helix syndrome: CLDN10
HSN: IV; VI
Idiopathic orthostatic hypotension
LEMS
Neuronal intranuclear inclusion disease
Pandysautonomia
Sjögren
Tangier disease
Tellurium
Pre- or Post-
ganglionicEctodermal
dysplasiaAnhidrotic
Recessive
ED + Immunodeficiency: IKBKG
ED: EDARADD
ED: EDAR
Dominant
ED + T-cell deficiency: NFKBIA
X-linked
ED1: EDA
ED: Other
Hidrotic
ED2: GJB6; 13q12; Dominant
Preganglionic Focal Diabetic radiculo-neuropathy
Leprosy
Local neuropathy (ulnar)
Horner's: + Ptosis; Miosis
Ross: + Hyporeflexia; Tonic pupils
SympathectomyPost-ganglionic Hemianhidrosis Cerebral infarction
Spinal cord tumorSympathetic outflow
CentralSkin disease Dermatophytosis
Limb-Mammary syndrome
Sweat ducts plugged
& Other
- Drugs
- Ach release inhibition: Presynaptic
- NMJ blocking agents: Botulinum toxin
- Anti-cholinergic: M3 receptor antagonism
- Anti-cholinergics: Atropine, Cyproheptadine, Doxepin, Glycopurrolate, Hyoscyamine
- Tricyclic antidepressant: Amitriptyline
- Anti-histamine: Diphenhydramine
- Anti-spasmodic, bladder: Oxybutinin, Toloterodine
- Anti-psychotics: Chlorpromazine, Clozapine, Quetiapine
- Carbonic anhydrase inhibition
- Anti-seizure: Topiramate, Zonisamide
- Central adrenergic
- Anti-hypertensive: Clonidine
- Hypothalamic μ-Opioid receptor antagonist
- Opioid: Fentanyl, Hydrocodone, Morphine, Oxycodone
- Ach release inhibition: Presynaptic
- Hereditary
- Anhidrotic ectodermal dysplasia (ED1)
● Ectodysplasin-A (EDA);
Chromosome Xq13.1 Recessive
- Protein: Small, 135 AA, transmembrane
- Clinical
- Hair: Sparse
- Teeth: Abnormal or missing
- Anhidrosis: Due to lack of sweat glands
- Hyperthermia: Life threatening or brain damage
- Carrier females: Abnormal teeth
- Hypohidrotic ectodermal dysplasia with immunodeficiency
● IKK-γ (IKBKG; NEMO); Chromosome Xq28; Recessive
- Genetics
- Mutations produce partial loss of IKK-γ function
- Allelic with incontinentia pigmenti
: Complete loss of IKK-γ function
- IKK-γ protein
- Required for activation of nuclear factor κ B transcription factor
- Role in T & B cell function
- Required for activation of nuclear factor κ B transcription factor
- Clinical
- Onset: < 2 years; Infections
- Hypohidrosis
- Abnormal dentition
- Osteopetrosis
- Immunodeficiency
- Low IgG
- High IgM or IgA
- Frequent infections
- Prognosis: Often early death
- Genetics
- Anhidrotic ectodermal dysplasia 11
ECTD11A: Dominant
ECTD11B: Recessive
● EDAR-associated death domain (EDARADD); Chromosome 1q42-q43
- EDARADD protein
- Expressed in epithelial cells during the formation of hair follicles and teeth
- EDARADD self-associates
- Death domain protein
- Interacts with death domain of EDAR
- Links the receptor to downstream signaling pathways
- Interacts with death domain of EDAR
- Clinical: Similar to X-linked AED
- Mouse: 'Crinkled' phenotype
- EDARADD protein
- Anhidrotic ectodermal dysplasia 10
ECT10A: Dominant
ECT10B: Recessive
● Ectodysplasin 1, anhidrotic receptor (EDAR);
Chromosome 2q12.3
- EDAR protein
- Structural similarity to TNF
- Triggers NF-kappa-B (see 164011) through the NEMO protein
- Clinical
- Similar to X-linked AED
- Abnormal morphogenesis of teeth, hair & eccrine sweat glands
- EDAR protein
- Anhidrotic ectodermal dysplasia + T-cell immunodificiency
● Nuclear factor of kappa light chain gene enhancer in B-cells inhibitor-α; (NFKBIA);
Chromosome 14q13.2; Dominant
- Mutations
- Heterozygous or Stop
- NFKBIA protein
- Inhibits NFKB complex
- Inactivate NF-kappa-B by trapping it in cytoplasm
- Clinical
- Multiple infections
- Conical teeth
- Skin: Dry; Rough
- Laboratory
- Skin biopsy: Absent sweat glands
- Mutations
- Anhidrotic ectodermal dysplasia: Other
● with Deafness
● with Retardation
- Hidrotic ectodermal dysplasia (ED2)
● Gap junction protein β-6 (GJB6, Connexin-30, CX30);
Chromosome 13q12.11; Dominant
- Anhidrosis, isolated, with normal sweat glands (ANHD)
● Inositol 1,4,5-triphosphate receptor, type 2 (ITPR2; InsP3R2);
Chromosome 12p12.1-p11.2; Recessive
- Nosology: Dann-Epstein-Sohar syndrome
- Epidemiology: Pakistani family
- Genetics
- Mutation: Homozygous; Gly2498Ser
- Also: ALS modifier locus
- ITPR2 protein
- Type 2 inositol 1,4,5-trisphosphate receptor (InsP3R2)
- Calcium ion transmembrane transporter activity
- Inositol 1,4,5-trisphosphate-sensitive calcium-release channel activity
- Phosphatidylinositol binding
- Membrane: Endoplasmic reticulum
- Downregulated by BMAL1
- Clinical
- Heat intolerance
- Anhidrosis: Generalized
- Laboratory
- Pathology, Sweat glands: Normal
- Eccrine sweating: Reduced
- HELIX Syndrome
● Claudin 10 (CLDN10);
Chromosome 13q32.1; Recessive
- Nosology
Hypohidrosis
Electrolyte imbalance
Lacrimal gland dysfunction
Ichthyosis
Xerostomia
- Epidemiology: 4 families; 19 patients
- Genetics
- Mutations: Missense; Met1Thr, Asn48Lys, Ser131Leu
- CLDN10 protein
- Expressed in: Dermis & Sweat glands: Renal
- Localize to: Tight junctions between epithelial cells
- Regulate: Transepithelial ion exchange; Electrical resistance
- Clinical
- Onset: Congenital
- Heat intolerance
- Anhidrosis
- Alacrima
- Xerostomia
- Nephrolithiasis: Onset in adolescence
- Pain: Recurrent bouts
- Dental: Enamel wear
- Laboratory
- Parathyroid hormone: High levels
- 25-Hydroxy vitamin D: Low
- Mg++ & Ca++: Low in urine
- Mg++: High in serum
- Plasma renin: High
- Sweat gland morphology: Normal
- Nosology
- Anhidrotic ectodermal dysplasia (ED1)
- General: Due to damage to
Urinary
- Nocturia: Postural hypotension
- Relocation of fluid from periphery
- Treatment: Desmopressin
- Nocturia: Cholinergic dysautonomia
- Bladder atony
- Common in Shy-Drager (Multiple System Atrophy): Lesion in sacral Onuf's nucleus
- Treatment
- Scheduled voiding with double voiding techniques
- Clean, intermittent self-catheterization
- Infection prevention
- Scheduled voiding with double voiding techniques
- Hereditary
Sexual dysfunction
- Erection: Dependent on parasympathetic supply & Nitric oxide
- Treatment
- Intracavernous injections of
- Prostaglandin E, or Papaverine ± Phentolamine
- Prostaglandin E, or Papaverine ± Phentolamine
- Topical nitroglycerin paste
- Physical: Surgery
- Intracavernous injections of
- Treatment
- Ejaculation: Dependent on sympathetic system
Ocular
|
Adie Horner's Ross Muscarinic AChR disorders Actin smooth muscle disorders Other pupil disorders Congenital MG Acetylcholinesterase (AChE) deficiency Hereditary neuropathy 2J (P0 mutations) HSAN 2B Miosis Tubular aggregate myopathy (Stormorken) TAM1: STIM1 TAM2: ORAI1 |
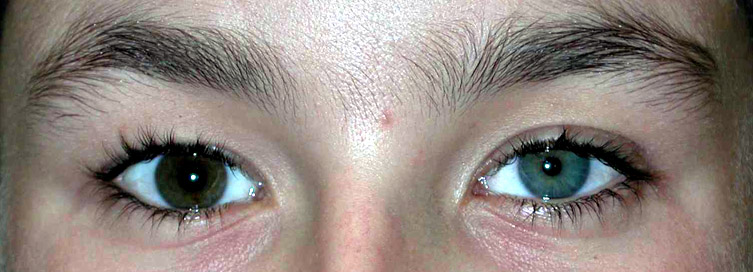 From: Monique Ryan Congenital Horner's: Iris heterochromia
|
- Horner's syndrome

From: P Bailey- Clinical features
- Ptosis
- Partial: Never covers visual field
- May affect upper (Müller's muscle) & lower lid (elevated)
- Pupil
- Miosis
- More evident in dark
- Never severe
- Dilation: Slow; Poor in dark
- Differential diagnosis of very small pupil
- Miotic drops
- CNS disorder
- Adie
- Argyll Robertson
- Miosis
- Hypohidrosis
- Preganglionic lesion proximal to carotid bifurcation
- Distribution: Face & Head
- Hyperemia: Conjunctival & lid; With acute lesion
- Facial vasodilation: Variable
- Enophthalmos: Appearance but rarely true
- Ptosis
- Lesions & Anatomy
- General anatomy: Ipsilateral sympathetic pathways
- CNS locations: "First order neurons"
- Anatomy
- Origin: Posterolateral hypothalamus
- Descending Pathway: Brainstem; Intermediolateral column of spinal cord
- Termination: Ciliospinal center (of Budge) in spinal cord at C-8 to T-2 levels
- Disorders
- Brainstem: Lateral medullary syndrome
- Cervical spinal cord: Disc; Osteophyte; Mass
- Child: Arnold-Chiari; Syringomyelia
- Anatomy
- Preganglionic: "Second order neurons"
- Anatomy
- Origin: Ciliospinal center (of Budge) of thoracic spinal cord
- Pathway: Mediastinum; Pulmonary apex; Supraclavicular space
- Ascends through: Stellate & Middle cervical ganglia
- Termination: Superior cervical ganglion at level of carotid bifurcation & angle of jaw
- Lesions
- Often neoplasm
- Usually with brachial plexus involvement
- Early lesion → Hyperfunction with mydriasis & hyperhidrosis
- Often neoplasm
- Anatomy
- Postganglionic: "Third order neurons"
- Anatomy
- Origin: Superior cervical ganglion
- Ascending pathways
- Internal carotid artery (Adventitia): Into skull & Cavernous sinus
- Sympathetic fibers
- Enter orbit through suprior orbital fissure
- Nerves: Long ciliary nerves
- Nasociliary branch of ophthalmic division of trigeminal nerve
- 2nd branch
- Passes through ciliary ganglion
- Joins postganglionic ciliary nerves (Short ciliary nerves)
- Termination
- Penetration of sclera
- Travel in suprachoroidal space
- Innervate: Dilator pupillae muscle; Inferior & Auperior (Müller) tarsal muscles
- Sympathetic fibers
- External carotid artery
- Vasomotor & Sudomotor innervation of hemiface
- Internal carotid artery (Adventitia): Into skull & Cavernous sinus
- Clinical Features
- Ptosis
- Miosis
- Pain
- ± Hypohidrosis
- Lesions
- Carotid artery dissection
- Intracranial (Cavernous sinus) or Internal carotid thrombosis
- Horner syndrome + VI nerve paresis: Ipsilateral posterior cavernous sinus
- Paratrigeminal syndrome: Pain in face & Horner's
- Anatomy
- Congenital Horner's
- Iris heterochromia: Blue iris on side of lesion
- Hereditary polyneuropathy
- Diagnostic testing
- Pharmacologic testing for localization: Compare responses in 2 eyes
- No pupil dilation by: Cocaine (10%) or Apraclonidine (0.5%)
- Postganglionic: No dilation with Hydroxyamphetamine (1%)
- Anatomic evaluation
- MRI with angiography; Brain & Cervical spine
- Pharmacologic testing for localization: Compare responses in 2 eyes
- Clinical features
- Holmes-Adie syndrome
- Hereditary
- Autosomal dominant
- HMSN: P0 mutations with axonal neuropathy
- Autosomal dominant
- Acquired
- Most common in young women
- Children: Often with history of chickenpox
- Specific causes
- Sjögren's
- Autonomic neuropathy
- Guillain-Barré: Occasional
- Pathology
- Parasympathetic, post-ganglionic, denervation
- Lesion: Ciliary ganglion; Posterior ciliary nerves
- Clinical features
- Pupil: "Tonic"
- Dilated
- Myotonic
- Light: Slowly or not reactive
- Near effort
- Strong & slow reaction
- Often tonic: Differs from Argyll Robertson
- Refixation at distance: Slow dilation
- Pathology: Reduced cells in parasympathetic ciliary ganglia
- Sectors of residual light reaction when viewed by slit lamp
- Unilateral or Bilateral
- Tendon reflexes: Areflexia
37
- H-reflexes may be preserved
- ? Due to pathology in dorsal root ganglia or posterior columns
- Other associated features
- Orthostatic hypotension
- Reduced Cardivascular reflexes; Syncope
- Hypo- or Hyper-hidrosis
- Carotid gustatory syndrome
- Diarrhea
- Chronic cough: Increased with deep breathing or hyperventilation
- Pupil: "Tonic"
- Hereditary
- Ross syndrome
13
- Epidemiology
- Males & Females
- > 80 patients
- Clinical features
- Onset age: 1st to 5th decade; Mean 36 years
- Anhidrosis
- Segmental or Generalized
- Residual sweating
- Most common on trunk
- Hyperhidrosis (compensatory) in these areas
- May be a main symptomatic feature with heat intolerance
- Course: Progressive loss; Areas may be hyperhidrotic before sweating loss
- Heat intolerance
- Hyporeflexia: Especially at ankles
- Tonic pupils (Adie)
- Slow reaction to light
- Normal constriction to a near target
- Dilute pilocarpine (0.062%) & adrenalin (0.1%): Postganglionic parasympathetic & sympathetic denervation of pupils
- Anisocoria
- Other dysfunction in some patients
- Cardiac
- Orthostatic hypotension
- Cough
- Sensory
- Subclinical changes
- Reduced temperature, pain & tactile sensation in some patients
- Course: May be progressive over years
- Electrophysiology
- H-reflex: Absent; Disorder of monosynaptic connection to motor neuron
- Nerve conduction: Normal
- Sympathetic
- Skin responses: Absent
- Peripheral outflow abnormalities
- Muscle sympathetic activity (MSNA): Spared
- Cardiovascular reflexes: Normal
- Pathology: Skin
- Post-ganglionic parasympathetic denervation: Sudomotor; Severe
- Sensory epidermal axon loss: Less severe
- Loss of unmyelinated & myelinated fibers
- Reduced innervation of sweat glands, blood vessels & erector pilorum muscles
- Adrenergic axon loss: Less severe
- Acquired disoders with similar features
- Sjögren syndrome
- Cholinergic urticaria
- ANA positivity
- Cytomegalovirus
- Hepatitis C, chronic
- Epidemiology
- Multisystemic Smooth Muscle Dysfunction Syndrome (MSMDS)
● Actin-α2, Smooth muscle, aorta (ACTA2); Chromosome 10q23.31; Dominant or Sporadic
- Epidemiology: 7 patients
- Genetics
- Mutations: Arg179His; Missense
- All de novo mutations
- Clinical
- Eye
- Mydriasis, Congenital
- Retina: Small vessel infarcts & aneurysms
- Cardiac & Vascular
- Patent ductus arteriosus
- Aortic aneurysm, thoracic (AAT6)
- Vasculopathy (Moya-Moya 5)
- Intestine
- Malrotation
- Hyperperistalsis
- Genitourinary
- Bladder: Hypotonic
- Cryptorchidism
- CNS
- Periventricular: White matter hyperintensities
- Small brain vessels: Brain infarcts & Aneurysms
- Eye
- Multisystem syndromes with congenital ptosis
Absence of lacrimation (Alacrima)
- Sjögren's
- LGMD R18
- Congenital absence
- Achalasia-Addisonianism-Alacrimia syndrome: AAAS
- Achalasia-Alacrimia-Intellectual Dysfunction: GMPPA
- Achalasia-Alacrimia-Peripheral Neuropathy: NDC1
- Alacrima congenita
● Autosomal Dominant
● Autosomal Recessive
- Riley-Day (HSAN 3): ELP1
- Congenital absence of lacrimal & salivary glands (ALSG)
● FGF10;
Chromosome 5p12; Dominant
- Allelic with: Lacrimoauriculodentodigital syndrome (LADD)
- Allelic with: Lacrimoauriculodentodigital syndrome (LADD)
- CDG1V: NGLY1
Respiratory disorders, Autonomic
- Congenital Central Hypoventilation Syndromes (CCHS)
\
General
CCHS
1: PHOX2B
2: MYO1H
3: LBX1
Other
- General
- Often associated with other autonomic disorders
- Hirschsprung: 30%
- Nosology: Ondine's curse
- Clinical criteria
- Hypoventilation, hypoxemia & hypercarbia during quiet sleep
- No cardiac, pulmonary, neuromuscular, EEG or cerebral MRI abnormality
- CCHS1
● Paired mesoderm homeo box 2B (PHOX2B; PMX2B); Chromosome 4p13; Dominant
- Most common gene mutated in congenital hypoventilation syndromes: 60% to 80%
- De novo mutations in 1st generation
- May be associated with Hirschsprung & Neoplasms
- Mutations
21
- Some patients with polyalanine expansion mutations
- Most common mutation type
- Usually in-frame duplication
- Add 5 to 13 alanines to normal 20-residue polyalanine tract
- Somatic mosaicism
- Mutation size same in different generations
- Late onset disease: 5 alanine insertion size
- Frameshift or Missense mutations: Many present with
- Hirschsprung disease
- Tumors of the sympathetic nervous system
- Types: Neuroblastoma, Ganglioneuroblastoma, Ganglioneuroma
- Location: Tumors often multifocal
- Some patients with polyalanine expansion mutations
- Disease mechanisms
- "Gain-of-function" alanine mutations: Cytoplasmic aggregation of normal & mutant proteins
- Haploinsufficiency mutations: Cause dilated pupils & ciliary ganglion atrophy
- CCHS2 & Autonomic dysfunction
● Myosin 1H (MYO1H); Chromosome 12q24.11; Recessive
- Epidemiology: 1 family, 3 patients
- Genetics
- Mutation: 1-bp Del, 2524A
- Myosin 1H protein
- Unconventional (Non-muscle) myosin
- Expression: Central nervous system
- Clinical
- Neonatal: Apneic spells; shallow breathing
- GI: Dysmotility; Poor swallowing
- Cardiac: Sinus bradycardia
- Temperature: Dysregulation
- Developmental delay: Global; Hypotonia
- Genetic associations: Other
● Endothelin-3;
Chromosome 20q13.2; Recessive or Dominant with reduced penetrance
- Also see: Hirschsprung
● RET oncogene;
Chromosome 10q11.21; Dominant or Sporadic
- Also see: Hirschsprung
- Malformation of enteric nervous system
● GDNF;
Chromosome 5p13.2; Dominant (HSCR1) or Recessive
- Also see: Hirschsprung
● BDNF;
Chromosome 11p14.1; Dominant
- Isolated CHS
- Father with postural hypotension & vasovagal syncope
● Human achaete-scute homolog-1 (HASH-1; ASCL1);
Chromosome 12q23.2; Dominant, Incomplete penetrance
- Mutations: Some are loss of alanines in polyalanine tract
18
- Normal: 13 Alanines
- Mutations: 5 or 8 Alanines
- HASH-1 protein expression
- CNS
- PNS
- Development: Enteric nervous system, Esophagus to rectum; Adrenal medulla
- Neuroblastoma
- HASH-1 Function
- Development of olfactory & most peripheral autonomic neurons
- Formation of neuronal circuits within CNS
- Mutations produce impaired noradrenergic neuronal development
- Interacts with E2-2 protein
- Clinical syndromes
- Central hypoventilation
- Haddad syndrome
- Central hypoventilation
- General
- Mental retardation + Intermittent hyperventilation (Pitt-Hopkins syndrome)
26,
57
● TCF-4
;
Chromosome 18q21.2; Dominant or Sporadic (de novo)
- Genetics: Mutations
- Heterozygous
- Types: Microdeletion; Missense; Stop; Splice site
- Allelic disorder: Corneal dystrophy, Fuchs endothelial, Dominant (FECD3)
- TCF4 protein
- E-protein: Class I basic helix-loop-helix transcription factor
- Subcellular location: Nuclear
- Expressed in fetal & adult brain
- Disease mechanism
- Haploinsufficiency
- ? Impaired noradrenergic & other neuronal development
- Complexes with ASCL1
- Clinical
- Onset: Infancy
- CNS
- Psychomotor delay: Severe
- Epilepsy
- Hyperventilation: Daily episodes; Diurnal
- Hypotonia
- May improve with pyridostigmine
- Morphology
- Growth retardation: Mild
- Microcephaly: Postnatal
- Face
- Nose: Large; High bridge; Flared nostril
- Mouth: Wide; Fleshy lips; Broad palate; Wide spaced teeth; M-shaped cupid’s bow
- Ears: Helices dysplastic & thick
- Eyes
- Enophthalmia
- Strabismus
- Eyebrows: Thin in midline portion
- GI: Some patients
- Hirschsprung disease
- Constipation
- ? Malignancy: Lymphoma in oldest patient
- Laboratory
- Repetitive Nerve Stimulation: Decrement
- Muscle: Type 1 fiber predominance (70%); TCF4 reduced
- CNS pathology
- Cerebellum: Hypoplasia of hemispheres & vermis
- Corpus callosum: Hypoplasia
- Hippocampus: Small
- Caudate nuclei: Bulging
- Genetics: Mutations
- Joubert syndromes
Multiple Systems Atrophy (Shy-Drager)
●
Sporadic or Autosomal Dominant
- Nosology: Other similar or overlapping disorders
- Striatonigral degeneration
- Adult-onset sporadic olivopontocerebellar atrophy (OPCA)
- Shy-Drager syndrome
- Genetic susceptibility
- Clinical features
- Onset
- Age: Adult
- Parkinsonism: Early
- Bladder dysfunction
- Orthostatic hypotension
36
- May initially present as: Pure autonomic failure
- Predictors of progression to MSA
- Cardiovagal impairment: Mild
- Sweat loss: Preganglionic pattern of sweat loss
- Bladder dysfunction: Severe
- Supine norepinephrine > 100 pg/mL
- Motor signs: Mild
- Autonomic
- Orthostatic hypotension
- Bladder incontinence
- Anhidrosis
- Ptosis
- Bowel dysfunction
- Extrapyramidal
- Parkinsonism (87%): Especially Rigidity, Postural instability & Bradykinesia
- Dystonic response to L-DOPA
- Upper motor neuron signs (50%): Spasticity; Brisk tendon reflexes
- Bulbar
- Scanning speech
- Stridor: Laryngeal abductor paralysis (also see Recessive ataxia)
- Cerebellar disorders (Especially OPCA variant): 50%
- Myoclonus: Stimulus-sensitive; Limbs & Face
- Peripheral nervous system
- Lower motor neuron signs (20%)
- Polyneuropathy: Sensory-Motor (20%)
- Skin: Dusky discoloration of the extremities
- Course
- Variably progressive
- Median survival: 6 to 10 years
- Onset
- Laboratory
- Bladder dysfunction (> 90%)
- EMG: Denervation (20%)
- Pathology
- Neuronal loss & Gliosis
- Extrapyramidal: Putamen; Substantia nigra; Locus ceruleus
- Other brainstem: Inferior olive; Pontine nuclei
- Purkinje cells
- Spinal cord: Intermediolateral cell column (Thoracic); Onuf's nucleus
- Glial cytoplasmic inclusions (Argyrophilic): Suprasegmental motor systems; Supraspinal autonomic system
- Autonomic pathology
- Spinal cord: Reduced sympathetic preganglionic neurons in intermediolateral cell column
- Medulla
- Reduced Catecholaminergic neurons (A1/C1) in ventrolateral intermediate reticular formation
- Normal projections
- Descending: Intermediolateral cell column in spinal cord
- Ascending: Vasopressin neurons in hypothalamus
- Neuronal loss & Gliosis
- External link: eNeurology
Riley-Day (HSAN 3)
19
●
Elongator complex protein 1 (IKBKAP; ELP1)
- Epidemiology: Most patients Ashkenazi-Jewish
- Genetics: IKBKAP mutations
- T to C transition in base pair 6 in donor splice site of intron 20 (2507+6T-C)
- Major haplotype: Present in 99.5% of disease alleles
- Common ancestral haplotype: Founder effect
- Carrier frequency in Ashkenazi-Jewish population: 1/36
- Mutation effect
- Variable skipping of exon 20
- Wild type message expressed in tissue-specific manner
- Lymphocytes: Mostly wild type protein
- Brain: Primarily mutant protein
- Arg696Pro: Missense mutation
- Rare
- All patients heterozygous for major haplotype
- Mutation predicted to disrupt potential threonine phosphorylation site
- Associated with mild phenotype
- Pro914Leu
14
- Found in non-Jewish patient
- Otherwise typical clinical syndrome
- Mutation disrupts protein phosphorylation
- Allelic disorder
- Medulloblastoma
- Medulloblastoma
- T to C transition in base pair 6 in donor splice site of intron 20 (2507+6T-C)
- IKBKAP protein
- Nosology: Inhibitor of κ light polypeptide gene enhancer in B cells, kinase complex-associated protein
- Member of complex containing other unidentified proteins
- Location: Highest levels of IKBKAP protein in cerebellum, thalamus, pituitary, testes
- Function: ? Role in gene-activation mechanisms
- Regulation & Activation of stress response through c-Jun N-terminal kinase (JNK) signaling path
- Mutant: May alter interaction of IKAP with JNK
- Misregulation of JNK
- Inadequate development & differentiation
- Mutant gene variably expressed
- Most of mRNA in brain is missing exon 20
- Wild type mRNA more abundant in fibroblasts
- Complete absence of expression in all tissues may be lethal
- Clinical features
- Onset: Congenital
- Early in disease course
- Newborn: Hypothermia; Vomiting crises; Hypotonia
- Eye
- Lacrimal Flow (Overflow tears): Reduced or Absent (99%)
- Cornea: Hypesthesia & Erosions
- Fungiform Tongue Papillae: Absent
- Sensory loss
- Modalities
- Pain Insensitivity
- Temperature: Abnormal Warm & Cold threshholds
- Functional muscle spindle afferents: Absent
- Large fiber modalities: Relatively spared
- Visceral sensation: Preserved
- Distribution: Trunk & Lower extremities most affected
- Modalities
- Deep tendon reflexes: Absent
- Autonomic crises
- General: Irritability, Fever, fainting
- Postural hypotension: No compensatory tachycardia
- Crises
- Triggers: Emotional or pysical stress
- Hypertension
- Tachycardia
- Hyperhidrosis
- Behavioral changes
- Treatments
- Vomiting: Diazepam
- Hypertension: Clonidine
- Skin blotching (99%)
- Hyperhidrosis (99%)
- Normal sympathetic skin responses
- GI disorders
- GI dysfunction: Dysmotility
- Esophageal & Gastric
- Dysphagia
- Gastroesophageal reflux
- Vomiting crises
- Skeletal
- Scoliosis
- Juvenile
- Left curves more common than idiopathic scoliosis
- Trauma: Repeated
- Fractures & Aseptic necrosis
- Neurogenic (Charcot) joints
- Scoliosis
- Respiratory
- Abnormal responses to hypoxic & hypercarbic states
- Aspiration: Recurrent pneumonias
- Restrictive lung disease
- Sleep: Central apnea & hypopnea
- Breath holding
- Cardiovascular: Postural hypotension
- Renal: Ischemia; Pre-renal azotemia
- Muscle: Possible increased frequency of rhabdomyolysis
35
- Frequency: 7.5 per 10,000 person-years
- Control: 0.44 per 10,000 person-years (Statin using patients)
- CNS: Normal intellect
- Course
- Older patients
- Finger nail dystrophy
- Taste: Reduced, especially sweet
- Vibration loss: With increasing age (> 13 years)
- Ataxia: Gait disorder; Poor balance
- Psychiatric syndromes
- Usually fatal: Death in 50% < 30 to 40 years
- Causes of death: Pulmonary; Renal; Sudden death
- Older patients
- Other
- Features specific to RD syndrome
- Lack of overflow tears
- Early onset postural hypotension
- Other Ashkenazi Jewish disorders
- Cystic fibrosis
- Tay-Sachs
- Canavan disease
- Features specific to RD syndrome
HSAN 3: Clinical Diagnosis
Overflow emotional tears Absent Lingual fungiform papillae Absent Patellar tendon reflexes Absent Axon flare after intradermal histamine Absent Ashkenazi Jewish heritage Present - Laboratory
- Hyponatremia: During crises
- Intradermal histamine: Absent flare
- Catecholamines
- HVA/VMA ratios: High
- DOPA/DHPG ratios: High in plasma
- Standing: Low increase in plasma levels of NE and dopamine β-hydroxylase
- Emotional crises: Very high plasma NE and DA levels
- Pathology
- Unmyelinated axons
- Number: Marked reduction
- 5% to 15% of normal
- Course: Progresasive loss
- Number: Marked reduction
- Dorsal root ganglia cell bodies: Reduced number
- Autonomic neurons: Loss
- Small superior cervical sympathetic ganglia
- Parasympathetic ganglia: Generally normal; Exception is sphenopalatine ganglion
- Large axons: Relatively spared: 65% to 100% of normal
- Spinal cord
- Reduced primary substance P axons in substantia gelatinosa
- Intermediolateral gray columns: Reduced neurons
- Unmyelinated axons
Congenital Sensory Neuropathy with Anhidrosis (HSAN 4; CIPA)
●
TRKA/ NGF receptor (NTRK1)
- Nosology: Congenital insensitivity to pain with anhidrosis (CIPA)
- Epidemiology: 1:25,000 incidence
- Gene mutations
- Types: Deletions, splice-site, frame shift, & missense
- > 53 identified: Most are private
- Domains: Signal peptide, extracellular & intracellular
- Geography
- High prevalence of disease in Israeli-Bedouin Arabs: Due to 1926-ins-T mutation
- Asian hot spot: c.851-33T>A
- Allelic disorder: Medullary thyroid carcinoma, familial
- Functional consequenses of mutations
- Extracellular domain: May prevent nerve growth factor binding to TRKA receptor
- Intracellular domain: Can interfere with signal transduction
- Protein
- Single extracellular, transmembrane & intracellular domains
- Extracellular domain: NGF binding; Signal peptide
- Intracellular domain: Tyrosine kinase
- Clinical features
- General
- Most: Homogeneous features
- Some: Incomplete syndromes 62
- Onset
- Congenital: Hypotonia
- Childhood: Delayed motor milestones; Hypohidrosis; Painless fractures
- Sensation
- Congenital insensitivity to pain
- Eye
- Decreased corneal sensitivity & reflexes
- Emotional tearing: Present
- Sensation: Reduced pain, temperature & visceral
- Some regional partial sparing of pain sensation
- Nose bridge
- Ears: Behind ears; External auditory meatus
- Posterior cervical skin area: Some patients
- Eye
- Temperature sensation: Absent or Reduced
- Normal: Touch, Vibration, Joint position
- Congenital insensitivity to pain
- Tendon reflexes: Normal, Increased or Decreased
- Autonomic
- Skin: Absent sweating (Anhidrosis); Blotching
- Normal: Blood pressure control; GI motility; Overflow tears
- CNS
- Mental retardation: Most patients
- Self mutilation (Acromutilation)
- Joint deformities: Knees & Ankles
- Mouth: Tip of tongue; Lips
- Skin: Scarring & Burns
- Systemic
- Fever
- Idiopathic
- Episodic & Recurrent
- Early disease manifestation: 3 months
- Episodic hyperpnea
- Fever
- Finger nail dystrophy
- Wound healing: Slow
- Course
- Early death from hyperpyrexia: Up to 20%
- Septicemia
- General
- Laboratory
- Provocative tests
8
- Histamine: Wheal evoked; No axon flare response (100%)
- Mecholyl: No tearing
- Pilocarpine: No sweating
- Sympathetic skin responses: Absent (100%)
- Plasma norepinephrine levels: Low or absent, Supine & Upright
- Nerve conduction velocities: Normal
- Anemia (79%)
- Provocative tests
8
- Pathology
- Peripheral nerve
- Small myelinated & Unmyelinated axons: Absent; 0% to 5% of noraml
- Large myelinated axons: Mildly reduced; 45% to 65% of normal
- Dorsal root ganglia
- Small sensory neurons absent: Associated with insensitivity to pain
- Sweat glands: Absence of innervation of eccrine glands
- Hypotrophic or absent in dermis
- Associated with anhidrosis & hyperthermia
- Autonomic
34
- Postganglionic sympathetic neurons: Probably severely depleted
- Chromaffin cells of adrenal medulla: Spared
- Skin: Reduced innervation; Remaining axons stain for GAP-43
- Peripheral nerve
- Variant syndrome: Milder phenotype
31
- NTRK1 Genetics
- Inheritance: Recessive
- Mutations
- c.851-33T>A: Splice site; Common in Korean patients
- Pro768Leu: May be associated with milder phenotype
- Clinical
- Skeletal: Recurrent bone fractures; Painless joint destruction
- Intelligence: Normal
- Motor: Strength normal
- Sensory exam: Vibration & Pin normal
- Laboratory
- EMG & NCV: Normal
- Sympathetic skin responses: Reduced
- Heart rate variance: Normal
- NTRK1 Genetics
HSAN 6: Autonomic Sensory Neuropathy
28
●
Dystonin (DST; BPAG1)
- Epidemiology: > 15 families; Ashkenazi Jewish
- Genetics
- Mutations
- Types
- Stop: c.14,865 delA (Ashkenazi)
- Missense
- Location: Neuronal DST-a isoform
- Types
- Allelic disorders
- Epidermolysis bullosa simplex, Autosomal recessive 2 (EBS3)
: Epithelial DST isoform (DST-e) mutations
- Axonal Neuropathy, Recessive
- HSAN 6: DST-a isoform mutations
- Arthrogryposis, Neurogenic (LCCS12): DST-a + DST-b isoform mutations
- Congenital Myopathy 29: DST-b isoform mutations
- Animal model: Dystonia musculorum (dt)
- Epidermolysis bullosa simplex, Autosomal recessive 2 (EBS3)
- Mutations
- Dystonin protein
- Cytoskeleton linker
- Actin filaments to Intermediate filaments & Microtubules
- Cytoskeleton organization during axonogenesis
- Hemidesmosomes: Isoform 3
- Axon transport, Retrograde
- Interactions: TMEM108; DCTN1
- Plakin family
- Size: Very large
- Alternatively spliced
- Creates many plakin family linker proteins
- Bullous pemphigoid antigen 1 (BPAG1) proteins
- Different isoforms in: CNS (DST-a); Muscle (DST-b); Skin (DST-e)
- Bundling actin filaments around nucleus: Nuclear envelope; Isoform 6
- Cytoskeleton linker
- Clinical
- Onset
- Age: Birth to 4th decade
- Hypotonia
- Respiratory failure
- Dysautonomia
- Absent tears
- Skin blotching
- GI dysmotility
- Feeding difficulties
- Diarrhea, chronic
- Hypohidrosis
- Episodic
- Hyperpyrexia
- Desaturation
- Bradycardia despite
- Blood pressure lability
- Tongue: Fungiform papilla Reduced
- Sensory loss
- Severe
- Distal
- Arms & Legs
- Panmodal
- Deep tendon reflexes: Absent
- Weakness: Intrinsic foot muscles
- Contractures: Distal arthrogryposis; Club feet; Hips; Knees; Fingers
- Osteomyelitis/Amputation: Distal; Hands or Feet
- Cranial
- Face: Motionless; Open-mouthed; Chin small
- Ears: Low set
- Palate: High arched
- Retardation (Ashkenazi family): Psycho-motor; Severe
- Course
- Ashkenazi: Early death (< 2 years)
- Italian family: Survival to adulthood
- Onset
- Laboratory
- Histamine test: Abnormal; No axon flare
- Skin biopsy: Reduced axon numbers
- Brain MRI: Normal
- DST variant: Axonal Neuropathy, Recessive
44
- Epidemiology: 1 family; 2 siblings
- Genetics
- Mutations
- Nonsense: p.R84X
- Affects 2 isoform variants with N-terminal transmembrane domain
- Splice donor: c.8283+1G>A; 22 amino acid in-frame deletion
- Location: Spectrin repeat domain of BPAG1a & BPAG1b isoforms
- Nonsense: p.R84X
- Loss of BPAG1 a2 & b2 isoforms: Correlates with axonal neuropathy
- Allelic disorders
- Mutations
- DST/BPAG1: Protein mutations
- Loss may disrupt axonal transport
- Actions via: Dynactinp150Glued 19; Retrolinkin; Microtubule-associated protein 1B
- Clinical
- Onset age: 2nd decade
- Weakness
- Distal
- Legs > Arms
- Sensory loss: Distal; Panmodal
- Tendon reflexes: Absent at ankles
- Progression: Gait disorder; Falling
- Ataxia
- Features: Dysmetria; Nystagmus; Scanning speech
- Onset: With disease progression
- Laboratory
- NCV: Axon loss, Motor & Sensory
- Brain MRI: Normal
- DST variant: LCCS 12, Arthrogryposis, Neurogenic
55
- Epidemiology: 5 patients
- Genetics
- Inheritance: Recessive
- Mutations
- Several gene domains
- May involve DST-a & DST-b
- Stop, Deletion, Missense
- Clinical
- Onset: Congenital
- Hypotonia
- Respiratory distress
- Face: Dysmorphic
- Arthrogryposis: Distal; Arms & Legs; Severe
- Severe phenotype: Early death in some
- Laboratory
- Nerve: Axon loss; Myelin sheaths thin
- Muscle: Normal
- DST variant: CMYO 29, Congenital Myopathy + Cardiomyopathy
60
- Epidemiology: 19 patients; 14 families
- Genetics
- Inheritance: Recessive
- Mutations
- Location: Exons 40-41; DST-b isoform
- Type: Mostly stop
- DST-b: Transcripts expressed in skeletal muscle, heart, fibroblasts
- Clinical
- Onset age: Neonatal or Prenatal
- Fetal movements: Reduced (50%)
- Arthrogryposis
- Hypotonia & Motor delay
- Respiratory insufficiency
- Dysphagia (50%)
- Cardiomyopathy, dilated
- Cognition: Normal
- Course
- Early death < 3 years: 50%
- Survivors: Improvement
- Laboratory
- Muscle
- Mildly myopathic
- Myofibrillar disruption mild
- Nuclear membranes undulating
- Lipid droplets
- Serum CK: Normal
- EMG: Myopathic
- Muscle MRI
- Hamstrings: Lipoatrophy
- Glutei: Atrophy
- Muscle
Parkinson disease, juvenile onset
●
Parkin
- Genetics: Mutations
- Location: Exons 2 to 9; Most frequent in exon 7
- Missense & small deletions
- Patients often compound heterozygotes
- Onset: Several syndromes
17
- Age
- Range: 7 to 64 years
- Usually: < 40 years
- Late: 1 patient with consanguinous parents
- Clinical syndromes at onset
- Parkinson syndrome: Tremor
- Dystonia: Exercise induced or Cervical
- Peripheral
- Autonomic dysfunction: may be present alone in 60%
- Peripheral neuropathy: Axonal
- Age
- Clinical
- Parkinson's: Bradykinesia; Rigidity; Akinesia
- Tremor: Leg & Hand
- Dystonia: Especially feet
- Dyskinesias: With treatment; 100%
- Tendon reflexes: Usually normal; Occasionally brisk
- Autonomic (60%)
- Urgency, urinary: 45%
- Impotence: 28% of males
- Othostatic symptoms: 13%
- Cortical
- Psychosis
- Cognitive function: Normal
- Course: Slow progression
- Treatment response: L-DOPA; Anticholinergics
Dopamine β-Hydroxylase deficiency (ORTHYP1)
41
●
Dopamine β-hydroxylase, plasma (DBH)
- Epidemiology: 21 patients
- Genetics
- Mutations: Missense & Splice
- DBH protein
- Function: Catalyzes oxidative hydroxylation of dopamine to norepinephrine
- Location: Adrenal medulla; Synaptic vesicles of postganglionic sympathetic neurons
- DBH axons
43
- Abundant around arterioles & arrector pili muscles
- Associated with: NPY
- Clinical
- Neonatal
- Eyes: Ptosis & Delayed opening
- Hypotension
- Hypoglycemia
- Hypothermia
- Childhood & Adult
- Postural hypotension
- Lightheadedness & Dizziness
- Syncope
- Progressive with exertion
- Nasal stuffiness
- Ejaculation: Prolonged or retrograde (normal erection)
- Exercise tolerance: Reduced
- Eyes
- Ptosis
- Intraocular pressure: Reduced with standing
- Pupils
- Size: Mildly small
- Respond to: Light & Accommodation; Parasympatholytics (Dilation)
- No response: Hydroxyamphetamine
- Sleep: Disordered
- Sweating: Normal
- Postural hypotension
- Exacerbation
- Heat
- Heavy meals
- Alcohol
- Skeletal
- Palate: High Arched
- Joints: Hyperextensible
- Cognition: Normal
- Treatment: L-threo-3,4-dihydroxyphenylserine (DOPS) - Elevates noradrenaline levels
- Dose: 100-600 mg (Droxidopa) po, 2x or 3x per day
- Neonatal
- Laboratory
- Dopamine beta-hydroxylase deficiency
- Noradrenaline & Adrenaline: Low in plasma, urine & CSF
- Dopamine: High in plasma, urine & CSF
- Autonomic function testing
- Systems
- Sympathetic: Failure
- Parasympathetic: Preserved
- Supine
- Blood pressure: Low to Normal
- Heart rate: Low to Normal
- Upright position assumed (or Valsalva maneuver)
- Systolic blood pressure: Falls < 80 mm Hg
- Compensatory rise in heart rate
- Inability to stand motionless for > Few minutes
- Hyperventilation: Blood pressue falls
- Systems
- Anemia (60%)
- BUN: Increased (67%)
- Glucose: Hypoglycemic episodes
- Skin biopsy: DβH axons lost; PGP 9.5 axons preserved 42
Orthostatic Hypotension 2 (ORTHYP2)
●
Cytochrome b561 (CYB561)
- Epidemiology: 2 families, 4 patients
- Genetics
- Mutations: Homozygous; Gly88Arg; Trp44X
- CTB561 protein
- Transmembrane protein
- Catecholamine & Neuropeptide secretory vesicles: Adrenal medulla, Pituitary, Neuroendocrine tissues
- Supplies reducing equivalents to
- DBH in chromaffin granules
- Peptidylglycine alpha-amidating monooxygenase (PAM)
in neurosecretory vesicles
- Expression in brain
- Clinical
- Onset age: Infant or Early childhood
- Orthostatic hypotension, severe
- Dizziness & occasional fainting on standing.
- No compensatory tachycardia
- Laboratory
- Sympathetic dysfunction
- Norepinephrine & Epinephrine & downstream metabolites: Decreased in plasma & urine
- Hypoglycemia: 1 family
- Anemia: 1 family
Dopamine receptor D4
● DRD4- Genetics
- Allelic disorder: Attention deficit-hyperactivity disorder (ADHD)
- Allelic disorder: Attention deficit-hyperactivity disorder (ADHD)
- Clinical features in 1 patient
- Autonomic hyperactivity
- Dermatographism
- Excess sweating
- Reduced body temperature
- Intermittent sinus tachycardia
- Dermatographism
- Acoustic neuromas
- Obesity
- Autonomic hyperactivity
- Novelty seeking personality trait
Orthostatic Hypotension, Streeten, Hereditary
●
Chromosome 18q; Dominant
- Nosology: Hyperbradykininism; Streeten type
- Genetics: Occasional non-penetrant patient
- Clinical
- Onset: Light-headedness on standing
- Ankle discoloration: Blue-purple
- Blood pressure
- Systolic: Reduced; Orthostatic drop in Systolic BP (>18 mm/Hg)
- Diastolic: Increased
- Tachycardia
- Signs & Symptoms resolve when patient is supine
- Progresses to syncope & palpitations
- Treatments: Propranolol, Fluorocortisone or Cyproheptadine
- Laboratory
- Plasma bradykinin: High
Aniridia, Type 2
●
Paired box gene 6 (PAX6)
- Allelic disorders
- Pax6
- Paired box gene family
- Transcriptional regulator involved in oculogenesis
- Governs cellular proliferation and migration of 'later-born' neurons
- Other tissues: Pancreas; Anterior pituitary
- Paired box gene family
- Clinical
- Eye
- Cataract
- Nystagmus
- Visual acuity: Reduced; Foveal hypoplasia; Glaucoma
- Blood pressure control: Abnormal
- Thermoregulation disorders
- Olfaction: Reduced
- Eye
- Other PAX6 syndromes
- Peters anomaly
- Ectopia pupillae
- Foveal hypoplasia
- Optic nerve hypoplasia or aplasia
- Keratitis
- Peters anomaly
- PAX6: 2 mutations
- Onset: Infancy
- Craniofacial: No eyes; Head small; Large ears; Nose flattened bridge, choanal atresia & pinpoint nares
- CNS: Absent optic nerves & chiasm; Brain small; Absent corpus callosum
- Death: Less than 1 month of age
- Other aniridia: Gillespie syndrome
- Animal model: Small eye mouse (Sey)
Achalasia-Addisonianism-Alacrimia (AAA) syndrome (Allgrove; AAAS; TAS)
11
●
AAAS (Aladin)
- Epidemiology
- Prevalence: 1 in 10,000
- Family history: Consanguinous families or Sporadic
- AAAS Gene
- Mutations
- > 75 described
- Location: All parts of gene
- Truncation, Frameshift, Nonsense, Splice & Missense
- ? Phenotype corelation: Compound heterozygotes
- One mutation is frameshift
- Second mutation
- Missense: Milder disease
- Stop or mutation in important protein region: More severe disease
- Associated with families having full AAA phenotype
- Allelic disorder: ALS-like syndrome
- Clinical AAA syndromes have genetic heterogeneity
- Mutations
- Aladin protein
- Type: WD-repeat family of regulatory proteins
- Gene expression
- Cerebral: ? Role in development of PNS & CNS
- Neuroendocrine: Adrenal gland
- Gastrointestinal tract
- Subcellular location: Nuclear
- Targeted to nuclear pore complexes (NPCs; Sites of nucleocytoplasmic transport)
- Functions
- Redox homeostasis
- Steroidogenesis in adrenal cells
- Mutated protein
- Common: Fails to localize to NPCs
- Impaired redox homeostasis & steroidogenesis
- Down-regulation: Cytochrome P450 hydroxylases (CYP17A1 & CYP21A2) & Cytochrome P450 oxidoreductase
- Reduction: DNA repair & Protection of cells against oxidative stress
- Other: Abnormal nuclear pore function
- Clinical
- Onset age
- Alacrimia at birth
- Achalasia & Adrenal: Childhood or Adult
- Neurological disorders: Later childhood or Adult
- GI
- Endocrine
- Adrenal insufficiency
- Glucocorticoid (85% to 90%)
- Mineralocorticoid (15%)
- Most with onset < 5 years
- Adrenal insufficiency
- Skin
- Hyperpigmentation
- Excessive creases on palms & soles
- Hyperkeratosis, Palmoplantar
- Skeletal
- Short stature
- Osteoporosis
- Microcephaly
- Dental carries
- Face dysmorphism
- Long thin
- Long philtrum
- Narrow upper lip
- Eyes
- Alacrimia
- Most consistent clinical feature
- Onset at birth
- Parasympathetic
- Optic atrophy
- Pupil disorders
- Pupillotonia
- Anisocoria
- Miosis
- Photophobia
- Alacrimia
- Peripheral neuropathy
- Modalities: Motor-Sensory or Motor
- Weakness: Distal; Legs & Hands
- Muscle Wasting: Distal
- Sensation: Often normal
- Pathology: Axon loss
- Tendon reflexes: Increased
- CNS
- Spasticity: Some patients
- Spinocerebellar syndrome: Ataxia; Dysarthria
- Optic atrophy
- Mental disability
- Deafness: Sensorineural
- Course: Progressive
- Autonomic dysfunction: General
- Parasympatheic & Sympathetic
- Cardiovascular
- Sudomotor: Dyshidrosis
- Hypotension
- Hyperhidrosis
From M Al-Lozi - Onset age
- Laboratory
- NCV
- Predominantly motor involvement
- Axonal loss (Small CMAPs); Mild slowing
- Sural: Reduced SNAP amplitudes with age
- Endocrine
- Hypoglycemic episodes: Seizures
- ACTH: High
- Dehydroepiandrosterone (DHEA-S) levels: Low (80%)
- Pathology
- NCV
- AAAS Variant syndrome: Adult onset, ALS-like disorder
27
- Clinical
- Onset age: Teens or Adults
- Bulbar
- Dysarthria
- Dysphagia
- Spastic tongue
- Weakness
- Onset: Legs
- Proximal & Distal
- Symmetric
- Motor neuropathy
- Distribution: Legs; Arms (Ulnar)
- Wasting: Hypothenar & other hand muscles; Calves
- Fasciculations: Limbs & Tongue
- Upper motor neuron
- Spasticity: All limbs
- Tendon reflexes: Brisk, except at ankles
- Autonomic
- Achalasia
- Dysautonomia
- Alacrimia
- Orthostatic hypotension
- Impotence
- Hyperhidrosis
- Endocrine: Adrenal insuficiency, later onset
- Other
- Lingua plicata
- Plantar hyperkeratosis
- Alacrimia
- Face: Hypertrophic nasal roots; Anterior cowlick
- Progression: Slow
- Laboratory
- NCV
- CMAP amplitudes: Small
- Normal velocity
- SNAPs: Normal
- EMG: Chronic denervation
- Brain MRI: Cerebral atrophy, moderate
- NCV
- Clinical
Achalasia-Alacrimia-Intellectual Dysfunction (AAMR)
32
●
Guanosine diphosphate (GDP)-mannose pyrophosphorylase A (GMPPA)
- Epidemiology: 14 families, 21 patients
- Genetics
- Mutations: Missense; Frameshift; Nonsense; Splice site
- GMPPA protein
- GMPPB
regulatory subunit
- Allosteric feedback inhibitor of GMPPB
- GDP-mannose: Component of N-glucan &, via dolichol-P-mannose, O-glycan chains in glycoproteins
- Mutation effects on α-Dystroglycan
48
- Enhanced mannose incorporation into glycoproteins
- Increased turnover of α-Dystroglycan
- Effect may be reversed by: Mannose restriction in diet
- Other CDGs in GDP-mannose pathway
- Ia (PMM2-CDG
)
- Ib (MPI-CDG
)
- Ia (PMM2-CDG
- GMPPB
- Clinical
- Onset age: Birth
- Achalasia: Birth to 7 years
- Alacrimia: Birth
- Intellectual disability
- Delayed milestones
- Speech delay
- Other autonomic
- Sweating reduction: 20%
- Postural hypotension: 30%
- Gait disorder: Some patients
- Hypotonia: Few patients
- Hyperkeratosis: 1 family
- Tendon reflexes: Usually normal
- Ocular
- Visual problems: Occasional
- Anisocoria: Some patients
- Hypotonia
- Skeletal
- Short stature
- Thumb anomalies: Hypoplasia
- Face dysmorphic: Columella Overhanging, Alae nasae hypoplastic, Mouth downturned, Philtrum short, Prognathia
- Adrenal: Normal
- Laboratory
- GDP-mannose levels (Lymphocytes): High
- Serum proteins: Normal N-glycosylation
- Muscle α-Dystroglycan
- Core protein: Reduced
- Glycosylation-specific epitopes: Increased
Neurodevelopmental Disorder with Achalasia-Alacrimia + Peripheral Neuropathy (NEDAPA)
56
●
NDC1 Transmembrane nucleoporin (NDC1; TMEM48)
- Epidemiology: 7 patients, 4 families
- Genetics
- Mutations: Missense or In-frame deletions
- NDC1 protein
- Anchor Nuclear pore complexes (NPC) to nuclear envelope
- Post-mitotic NPC assembly
- Recruit ALADIN to nuclear envelope
- Mutation: Decreased ALADIN in nuclear envelope
- Upregulation: Ischemic & Dilated cardiomyopathy
- Clinical
- Onset age: < 1 year
- CNS: Intellectual disability; Speech delay
- Motor: Impairment; Hypotonia
- Polyneuropathy
- Face: Weakness; Nasal speech
- Tongue: Fasciculations
- Alacrima: Few other autonomic features
- Achalasia: Feeding disorders; Dysphagia
- Skeletal
- Face dysmorphism
- Contractures: Few patients
- Fingers: Tapered
- Laboratory
- NCV
- Demyelination: Severe
- Axon loss
- Muscle CT: Gastrocnemius & Soleus atrophy
- Brain MRI: Normal or Ventricular dilatation
- NCV
Autoimmune polyendocrinopathy-Candidiasis-Ectodermal dystrophy (APECED)
●
Autoimmune regulator (AIRE)
- Nosology: Autoimmune polyendocrine syndrome, Type I (APS1) ± Reversible metaphyseal dysplasia
- Epidemiology: Mostly in Finns, Sardinians & Iranian Jews
- Genetics
- Mutations: > 60 described
- Allelic disorder: Myasthenia gravis susceptibility in Chinese
- AIRE protein
- Promotes T cell tolerance to self-antigens
- Upregulates ectopic expression of tissue specific self-antigens in medullary thymic epithelial cells (mTECs) in thymus
- Promotes deletion of T cells that recognize self-antigens with high affinity
- Mutation: Defective negative selection of self-reactive effector T cells
- Clinical
- Onset age: 1st decade
- Candidiasis: Chronic mucocutaneous
- Endocrine
- 1° Hypoparathyroidism
- 1° Hypogonadism
- Adrenal insufficiency
- Diabetes mellitus: Insulin-dependent; GAD antibodies
- Hypothyroidism: Latent
- Hypoaldosteronism: Transient, Isolated
- Gonadal failure: Primary sertoli cell insufficiency
- Autoimmune disorders, Multiple
- Myopathy
30
- Frequency: Few patients
- Clinical
- Onset age: 14 to 45 years
- Most patients female
- Weakness
- Proximal > Distal
- Arms & Legs
- Symmetric
- Head drop
- Respiratory: Some patients
- Atrophy: Proximal & Trunk
- Course: Slowly progressive to disability & respiratory failure
- Muscle pathology
- Myopathy: Varied muscle fiber size
- Internal architecture: Irregular
- Type 1 muscle fiber atrophy
- Inflammation: None
- MHC-1: Normal
- Laboratory
- Serum CK: Normal
- Neuropathy
- 2 adolescent males
- Clinical: Weakness; Sensory loss
- NCV: May have demyelination
- Hair: Alopecia totalis
- Nails: Dystrophy
- Teeth: Dental enamel hypoplasia
- Eyes: Keratopathy; Keratoconjunctivitis
- GI: Malabsorption; Diarrhea; Chronic active hepatitis; Achalasia
- Laboratory
- Pernicious anemia: Juvenile-onset
- Humoral immunity
- Hypogammaglobulinemia
- Antibodies: Antiadrenal, Antithyroid & Antinuclear
- Cellular immunity: Low T4/T8 cell ratio
- Mouse model: AIRE G228W mutation on autoimmuneprone non-obese diabetic (NOD) background
- Develop
- Spontaneous autoimmune polyneuropathy with demyelinating features
- P0 (MPZ) antibodies
- Develop
Postural Orthostatic Tachycardia Syndrome (POTS) 2
|
General Genetics Clinical Laboratory Treatment |
- Nosology: Postural Tachycardia Syndrome (PoTS)
- Definition
- Orthostatic intolerance, Symptomatic
- Standing: Worse
- Supine: Improved
- Associated with: Heart rate increment
- Rates
- ≥ 30 beats/minute, or
- ≥ 40 bpm if age ≶ 20 years, or
- > 120 bpm
- Association: Within 10 minutes of Δ from supine to upright
- No orthostatic hypotension
- Rates
- Symptom duration: ≥ 6 months
- No other cause found
- Orthostatic intolerance, Symptomatic
- Epidemiology
- 0.2% to 1% of US population
- Female: Male = 5:1
- Genetics: Family history in some patients
- Orthostatic intolerance
● SLC6A2; Chromosome 16q12.2; Dominant
- Epidemiology: 1 family; 2 identical twins
- Genetics
- Mutation: Missense Ala457Pro
- Variable penetrance
- SLC6A2 protein
- Norepinephrine transporter
- Mutated norepinephrine transporter protein: Non-functional
- Clincal features
- Onset age: 2nd decade
- Fatigue
- Orthostatic intolerance
- Altered heart-rate regulation
- Upright posture: Heart rate increases by ≥ 30 beats per minute; No orthostatic hypotension
- Syncope
- Laboratory: Altered norepinephrine metabolism
- Ehlers-Danlos, Type III (Hypermobility) (EDSHMB)
- Orthostatic intolerance
- Clinical
- Patient characteristics
- Young: Mean 14 to 29 years; Range 12 to 50 years
- Female to Male: 4:1
- Onset
- Course: Acute, Subacute, or Insidious
- Prodromes
- Febrile or Viral illness & other infections: 50% of patients
- Other: Trauma, Surgery Stress, Pregnancy
- Symptoms
- General: Associated with vaso-dilation in vascular beds
- Develop on standing
- Resolve with recumbency
- 3 or more symptoms present for > 3 months
- Weakness: Lower extremity or Diffuse
- Light-headedness or Dizziness
- Vision: Blurred
- Palpitations
- Dyspnea: Breathing difficulty
- Nausea
- Headache: Increased with standing
- Other symptoms
- Tremor
- Prodrome: Infectious (50%)
- Exercise intolerance: Fatigue
- Syncope
- Pain: Chest or Abdomen
- Dry eyes or mouth
- GI: Bloating; Early satiety; Diarrhea & Constipation
- Symptoms aggravated
- Heat
- Meals: Food or Alcohol
- Dehydration
- Exertion
- Time of day: Early
- Menstrual
- Speed of postural change
- Signs
- Heart rate
- Increase of > 30 bpm: Within 10 minutes of standing
- Increase usually to ≥ 120 bpm
- No orthostatic hypotension
- Venous pooling in legs with standing
- Sweating in legs: Reduced
- Joint hypermobility
- Mitral valve prolapse
- Heart rate
- Prognosis
- Improvement in majority (80%): 60% may become normal
- Better with history of infectious prodrome
- Associated syndromes
- Migraine headache
- Irritable bowel
- Mast cell activation
- Treatment
- Volume expansion
- Diet: High salt (In patients without hypertension)
- High fluid intake
- Rapid water drinking, 120 to 480 ml over 5 minutes
- Especially in morning
- Meals: Small; Frequent
- Head-up tilt: At night
- Physical countermaneuvers
- Elastic stockings & Abdominal binders
- Pharmacologic
- Fludrocortisone
- < 300 µg daily
- Not with: Fluid retention; Hypertension
- β-adrenergic antagonists
- Bisoprolol or Propranolol (20 mg)
- Low doses
- Contraindicated with asthma
- Midodrine: Sympathetic denervation
- 2.5 mg 3 times daily before meals
- Increase after few weeks if necessary
- Maximum dose: 30 mg daily
- Not used with hypertension
- Pyridostigmine: GI side effects common
- Octreotide
- For: Post-prandial symptoms
- Subcutaneous: 25 to 50 µg before food ingestion
- Ivabradine: Sinus node blockade; For more severe tachycardia
- Clonidine
- Fludrocortisone
- Volume expansion
- Patient characteristics
- Laboratory: Partial sympathetic denervation, especially in legs
5
- Tilt testing: Excessive heart rate increment
- Test: Tilt-up 60° for 10 to 60 minutes
- Outcome
- Tachycardia: 120 to 170 bpm on tilt-up
- Reach levels by by 2 min
- May increase progressively over time
- Blood pressure: No change
- Adrenergic testing
- Stimuli (Cold pressor; Na nitroprusside; Tyramine): Reduced increase of norepinephrine levels in legs
- Plasma norepinephrine levels
- Supine: Normal or Low (60% of normal)
- Standing: Hyperadrenergic subgroup; Rise to > 600 pg/ml
- Valsalva maneuver
- Moderately forceful attempted exhalation against a closed airway
- Impaired baroreflex-mediated peripheral vasoconstrictor function
- Cardiovagal tests: Normal
- Sweating: Distal hypohidrosis, postganglionic
- Skin biopsy: Abnormal in 24%; Neuropathic subgroup
- Possible serum autoantibodies: Adrenergic & Cholinergic receptors 58
- Tilt testing: Excessive heart rate increment
Hypotension in the Elderly 6
- Associated factors
- Standing: Hypotension increases with length of time standing
- Post-prandial
- High carbohydrate content of meal
- Warm temperatures
- Anti-hypertensive medications
- Clinical: May be associated with syncope
Adult-onset Leukodystrophy (ADLD)
3
●
Lamin B1 (LMNB1)
- Epidemiology: 33 families
- Genetics
25
- Mutation: Duplication including
- Lamin B1 gene
- MARCH3: 3' end
- Other regulatory sequences
- Allelic disorders
- Microcephaly 26, primary, autosomal dominant (MCPH26); de novo Missense mutations
- Atypical ADLD (ADLDAT)
61
- Mutation: Chromosome 5q23 deletion involving regulatory regions upstream of LMNB1 gene
- Clinical: Pyramidal before Autonomic; No cerebellar involvement
- Microcephaly 26, primary, autosomal dominant (MCPH26); de novo Missense mutations
- Mutation: Duplication including
- Lamin B1 protein
- Widely expressed
- Component of nuclear lamina (envelope)
- Mutation: Over-expression in brain
- Over-expression causes abnormal nuclear morphology: Blebbing & folding of membrane
- Clinical
- Onset
- Adult: 30's to 50's; Mean 41 years
- Autonomic features first
- Autonomic
- Bowel/bladder dysfunction
- Impotence (in males)
- Orthostatic hypotension
- Hypohidrosis
- Pyramidal
- Loss of fine motor skills
- Spastic paraparesis or tetraparesis ~90%
- Pseudobulbar palsy
- Posterior column dysfunction
- Cerebellar
- Nystagmus 69%
- Ataxia 100%
- Cognitive impairment: Some patients
- Course
- Slowly progressive over 20 years
- Loss of voluntary movement
- Fatal
- Onset
- Differential diagnosis
- Progressive multiple sclerosis
- Toxic demyelination: Hexachlorophene; Cuprizone
- Laboratory
- CNS MRI: Leukodystrophy
- Symmetric widespread myelin loss
- T2 hyperintense lesions
- Begins in frontal & parietal lobes
- Extends to cerebellum
- Spinal cord: Atrophy
- Neuropathology
- Locations: White matter of centrum semiovali & cerebellar peduncles
- Myelin loss in isolated and confluent patches
- Microscopic
- Vacuolation of white matter
- Oligodendrocytes abundant within lesions
- Astrocytes sparse: Processes abnormally beaded & foreshortened
- No inflammation or clear neuronal pathology
- CNS MRI: Leukodystrophy
Cold-Induced Sweating Syndrome
12
●
Cytokine receptor–like factor 1 (CRLF1)
- Epidemiology: Israeli & Norwegian families
- Norwegian family: Neonatal; Feeding & Respiratory disorders
- Gene mutations
- Norwegian family: More severe; 844_845delGT
- Israeli family: 2 missense mutations; R81H & L374R
- Also occur in: Crisponi syndrome
- CRLF1 protein
- Expressed in skeletal muscle & other tissues
- Soluble cytokine receptor: Homology to type 1 receptors
- Associates with cardiotrophin-like cytokine
- Competes with CNTF for binding to CNTF receptor
- Syndromes involving related proteins
- Clinical
- Onset age: Variable
- Sweating
- Onset: 1st or 2nd decade
- Induced at temperatures of 7°C to 18°C
- Anatomical distribution
- Sweating begins in localized region: Presternal; Hand
- Becomes profuse on large segments: Above waist; Patches on back and chest
- No sweating in affected areas with heat or fever
- Course: Non-progressive
- Sensory loss: Small fiber modalities
- In more severely affected patients
- Reduced heat & surgical induced pain
- Reduced sensation of heat in hot environments
- Feeding: Reduced interest in food
- Skeletal
- High-arched palate: Nasal voice
- Face
- Reduced facial expression
- Depressed nasal bridge
- Limited opening of mouth
- Elbow contractures
- Clinodactyly
- Kyphoscoliosis: Thoracolumbar
Cold-Induced Sweating Syndrome 2
●
Cardiotrophin-like cytokine factor 1 (CLCF1)
- Epidemiology: Single Australian patient
- Genetics: Heterozygous mutations; Tyr107Ter & Arg197Leu
- CLCF1 protein
- Family: Interleukin-6 cytokines
- Syndromes involving related proteins
- Clinical
- Onset: Infancy; Feeding difficulties; Sweating
- Sweating
- Induced by cold
- Inability to sweat in hot weather
- Location: Especially face, trunk & upper limbs
- Face
- Weakness: Mild
- Ears: Set at right angles to skull
- Polyneuropathy
- Predominantly sensory
- Axon loss
- Skeletal & Morphological
- Palate: High-arched palate
- Elbows: Valgus deformity; Inability to extend fully
- Clinodactyly: Fingers & Toes, syndactyly of second and third toes of both feet
- Thoracolumbar scoliosis
- Lmbar lordosis
- Degenerative disease of the cervical & lumbar spine
- Course: Non-progressive
Cold-Induced Sweating Syndrome 3/Crisopni-like) (CISS3)
●
Kelch-like 7 (KLHL7)
- Epidemiology: 4 Turkish families, 5 patients
- Genetics
- Mutations
- Homozygous
- Missense & Truncation: Arg420Cys, Cys421Ser, Arg372Gln, 1-BP del, 1022T
- In or near Kelch domain
- Allelic disorder: Retinitis pigmentosa 42
- Mutations
- KLHL7 protein
- Substrate recognition subunits for cullin-3
-containing E3 ubiquitin ligase complexes
- Polyubiquitinate target proteins for proteasome-mediated degradation
- Substrate recognition subunits for cullin-3
- Clinical: Crisponi-like
- Onset age: Infancy
- Hyperthermia
- Paroxysmal contractions of oropharyngeal muscles: Feeding & swallowing difficulties.
- Skeletal: Camptodactyly, Joint contractures, Full cheeks, High-arched palate, Depressed nasal bridge
- Cold induced sweating: 1 patient at 4 years
- Retinitis pigmentosa: After 2 years
- Course: Death at 7 to 24 months in 3 patients
Harlequin Syndrome
|
|
|
Neuronal Intranuclear (Hyaline) Inclusion Disease (NIID)
38
●
Notch2 N-terminal-like C (NOTCH2NLC)
- Epidemiology
- > 140 patients
- Asians: Most common cause of NIID
- Europeans: GGC repeat expansions unusual
- Genetics
- Mutations
- CGG (GGC) repeat expansions (66 to 517)
- Location: 5' untranslated region of gene
- Possible GGC repeat disease mechanisms
63
- GGC repeats are in open reading frame
- GGC repeat expansions are translated into new polyglycine-containing proteins
- Polyglycine proteins induce
- OPDM/OPML p62 inclusions
- Muscle fiber atrophy
- Allelic disorders
- Neuromuscular
- Oculopharyngodistal Myopathy + Neurological features (OPDM3)
- Polyneuropathy, Axonal (Motor CMT)
- ALS
45
- CGG mutations present in 0.7%
- Severe & Rapid disease
- Neurodegenerative
- Multiple system atrophy (MSA)
- Leukoencephalopathy
- Alzheimer’s disease + Frontotemporal dementia (AD/FTD)
- Tremor, essential 6
- Retinal dystrophy
- Parkinsonism: Intermediate length GGC expansion
- Neuromuscular
- Other 5' CGG expansion disorders
- Mutations
- NOTCH2NLC protein
- Brain: Expression increases with age
- Interacts with Notch receptors & enhanced Notch signaling
- Clinical
- Onset
- Age: Usually childhood
- Autonomic disorders
- Autonomic failure
- Incontinence: Fecal; Urinary
- Postural: Hypotension; Light-headedness; Syncope
- Erectile dysfunction
- Hypohidrosis
- GI: Pseudo-obstruction
- Course: May be progressive or episodic
- Cerebellar dysfunction
- Ataxia: Gait
- Dysarthria
- Ocular dysmetria
- Tremor
- Peripheral neuropathy: Some families
23
- Motor: Weakness of face & limbs; Nasal voice; Dysphagia
- Sensory loss: Pan-modal; Distal
- Electrophysiology: Axonal loss
- Nerve pathology
- Axon loss: Small > Large
- Schwann cell inclusions: Stain with ubiquitin
- Motor
- Weakness: Legs or Diffuse
- Hypotonia
- Tendon reflexes: Increased or Reduced (with neuropathy)
- Onset
- Laboratory
- MRI
- Cerebellar ± Cortical atrophy
- DWI signal intensity in corticomedullary junction of cerebral white matter
- EMG: Distal denervation; Sphincter abnormalities
- Muscle biopsy: Normal
- Serum CK: High in some patients
- Skin biopsy: Small fiber neuropathy, non-length-dependent, common 59
- MRI
- Pathology
- Neuronal & glial nuclear inclusions
- Eosinophilic
- Ubiquitinated
- Neuronal nuclear
- Locations
- Widespread in CNS & PNS
- Neuronal, Rectal or Skin biopsy (Ubiquitin staining)
- Peripheral nerve with sensory involvement
- Neuronal loss & gliosis
- Sympathetic & Sensory ganglion neurons
- Spinal & Brainstem motor neurons
- Neuronal & glial nuclear inclusions
- NOTCH2NLC variant: Oculopharyngodistal Myopathy + Neurological features
(OPDM3)
46
- Epidemiology: 7 Japanese & 5 Chinese patients
- Genetics
- Inheritance: de novo individuals or single generation
- Mutations: NOTCH2NLC CGG expansions, 116 to 674 repeats
- Clinical
- Onset age: Infancy to 67 years
- Eye: Ptosis, Ophthalmoplegia
- Weakness
- Bulbar: Dysarthria, Dysphagia
- Face
- Limbs: Often predominantly distal
- Deep tendon reflexes: Reduced
- Neural: Varied
- Leukoencephalopathy
- Retinal pigmentary degeneration
- Ataxia
- Tremor
- Deafness
- Peripheral neuropathy
- Laboratory
- Muscle pathology
- Fiber sizes: Varied
- Vacuoles, rimmed
- Immature muscle fibers: Myofibers with neonatal myosin heavy chain
- Intra-myonuclear inclusions: Poly-ubiquitinated proteins, SUMO1, Phospho-p62/SQSTM1
- Ultrastructure: Nuclear tubulofilamentous inclusions without limiting membrane
- Skin: p62-positive intranuclear inclusions
- Muscle imaging
- Asymmetric
- Muscles involved: Gluteus maximus, Vastus lateralis, Adductor magnus, Rectus femoris
- Brain MRI
- FLAIR: Middle cerebellar peduncles; Paravermal
- DWI: Corticomedullary junction
- Serum CK: 436 to 1886
- CSF protein: High
- Muscle pathology
- NOTCH2NLC variant: Polyneuropathy, Axonal or Intermediate
54
- Epidemiology
- 39 patients
- Geographic CMT Frequency: 6% to 10% of Taiwan, 3% China, 1.2% Japan
- Genetics
- Clinical
- Onset
- Age: Mean 34 to 37 years; Range 7 to 61 years
- Tingling: Feet or Hands
- Polyneuropathy
- General
- Often motor predominant
- Sensory only: Few patients
- Mild severity
- Sensory
- Paresthesias (72%): Legs, Arms, Tongue
- Pin or Vibration loss (57%)
- Cramps (43%)
- Weakness (43%)
- Distal > Diffuse
- Legs > Arms
- Pure motor PN: 1 patient
- Tendon reflexes: Reduced or Absent
- General
- Autonomic
- Abnormal (44%): Especially in more severe disease
- Neurogenic bladder
- Erectile dysfunction
- Anhidrosis
- Constipation
- Orthostatic hypotension
- CNS
- Involuntary movements (29%): Most Postural tremor
- Myoclonus (9%)
- Course: Slow progression
- Onset
- Laboratory
- NCV
- General: Intermediate range (69%); Axonal (31%)
- Mild slowing (Arms > 38 m/s)
- CMAP & SNAP amplitudes: Often normal
- Skin biopsy
- Inclusions: p62 & Ubiquitin
- Eosinophilic
- Location: Sweat gland cells; Dermal fibroblasts
- Serum CK: High in 75%; Mean 559; Range 51 to 1681
- Nerve biopsy
- Axon loss
- Thinly myelinated axons
- Ultrastructure: Intranuclear inclusions
- Filamentous aggregates
- In Schwann & perineurial cells
- Brain MRI: Atrophy or Luecoaraiosis
- NCV
- Epidemiology
Hypotonia, Hyperventilation, impaired Intellectual development, Dysautonomia, Epilepsy & Eye Abnormalities (HIDEA)
●
Prolyl 4-Hydroxylase, Transmembrane (P4HTM)
- Epidemiology: 5 families, 13 patients
- Genetics
- Mutations: Loss of function
- P4HTM protein
- Expression highest: Brain & Eye
- Oxygen-dependent regulation of hypoxia-inducible factor (HIF)
- Mutations: Insoluble proteins
- Clinical
- Onset age: 1 top 5 years
- Hypotonia
- Intellectual disability
- Eye: Strabismus
- Epilepsy
- Respiratory: Pneumonia, recurrent; Sleep apnea
- Autonomic: Constipation; Hypo- or Hyperthermia; Hypohidrosis
- BMI: High (50%)
- Laboratory
- Brain MRI: Normal
- Muscle biopsy
- Mitochondria: Complex activities reduced; Abnormal morpohology; COX- fibers
- Fiber sizes: Varied
Dementia with Lewy bodies
20
- Genetics: Mutations in
- SNCA
- SNCB
- SNCA
- Clinical
- Onset age: 65 years
- Cognitive
- Decline: Progressive
- Fluctuating: Cognition or Arousal
- Visual hallucinations: Recurrent, Formed
- Parkinsonism: Spontaneous
- Falls
- Syncopal episodes
- Sensitivity to neuroleptic medication
- Autonomic dysfunction
- Anhidrosis: Distal
- Hypotension, Orthostatic
- Urinary symptoms (35%)
- Brain pathology: Lewy bodies, widespread
Mast Cells
Mast Cells: General 51
|
Mast Cell
|
- Immune: Allergy, Asthma, Anaphylaxis
- Gastrointestinal disorders
- Malignancies
- Cardiovascular diseases
- Neuroinflammation
- Idiopathic mast cell activation syndrome (MCAS)
- Small fiber neuropathy (80%)
- Dysautonomia
- Serum tryptase: Baseline < 8 ng/ml
- Hereditary α-tryptasemia (HαT)
52
● Tryptase α/β-1 (TPSAB1; MCP7);
Chromosome 16p13.3; Dominant
- Epidemiology
- 5% to 7% of Western populations
- Greater in patients with known severe anaphylaxis
- Genetics
- TPSAB1: Gene copy number increased
- Clinical
- Cutaneous: Flushing & Pruritus
- Gastrointestinal: Lower GI symptoms
- Neurological
53
- Pain: Chronic
- Small fiber neuropathy (80%)
- Dysautonomia (100%)
- Sympathetic, Parasympathetic or Sudomotor
- Orthostatic intolerance
- Sweating disorders
- Cerebral blood flow
- Orthostatic: Reduced
- "Brain fog"
- Connective tissue: Primary dentition retained
- Anaphylaxis: Severe
- Asymptomatic: Many carriers
- Laboratory
- Serum tryptase: Baseline (BST) high
- > 8 ng/ml
- Additional TPSAB1 copy encoding α-tryptase: BST increased by 9 to 10 ng/mL
- Mast cells, Bone marrow: Increased, Atypical
- Serum tryptase: Baseline (BST) high
- Epidemiology
Return to Neuromuscular home page
Return to Polyneuropathy Index
Return to Autonomic physiology & Pharmacology
References
1. Neuropediatrics 1996;27:26-31
2. Mayo Clinic Proc 1999;74:1106-1110, Nat Rev Neurol 2011;8:22-34, Curr Probl Cardiol 2022;47:101384, Trends Cardiovasc Med 2023;33:65-69
3. Human Molecular Genetics 2000;9:787-793, Front Neurosci 2025:19:1531593
4. NEJM 2000;343:847-855, Arch Neurol 2009;66:735-741
5. NEJM 2000;343:1008-1014
6. Ann Int Med 2000;133:533-536
7. Muscle Nerve 2001;24:292-296
8. Pediatr Neurol 2001;25:397-400
9. Am J Med 2002;112:355-360
10. Nature Genetics 2002;May
11. Brain 2002;125:2681-2690, Endocrine 2023;79:376-383
12. Am J Hum Genet 2003;72:Online January
13. Acta Neurol Scand 2003;107:72–73, Clinical Neurophysiology 2003;114:7–16, Acta Neurol Taiwan 2023:32:127-130
14. Am J Med Genet 2003;Online March
15. J Clin Invest 2003;111:907–913, J Clin Invest 2003;111:797-799
16. Muscle Nerve 2003; Online February 2003; Neurology 2009;73:1501–1506
17. Brain 2003;126:1279-1292
18. Human Mol Genet 2003; Online October
19. Muscle Nerve 2004;29:352-363
20. Neurology 2004;62:1804–1809
21. Am J Hum Genet 2005;72:March
22. Am J Hum Genet 2005;77:Online May
23. Neurology 2005;65;1538-1543
24. Arch Neurol 2006 63:513-518
25. Nat Genet 2006 Sep 3
26. Am J Hum Genet 2007; Online Mar
27. Eur J Hum Genet 2008 Jul 16; Amyotroph Lateral Scler 2008 Jul 10:1-3
28. Ann Neurol 2012; On-Line Jan, Exp Dermatol 2022 Mar 11
29. Dermatology 2012; Jan 12
30. Muscle Nerve 2012;45:904–908
31. J Child Neurol 2012 Oct 30
32. Am J Human Genet 2013; Online Sept, Am J Med Genet A 2020;182:425-430
33. Neurology 2014;83:1-7, Neurol Neuroimmunol Neuroinflamm 2025;12:e200350
34. Ann Neurol 2015;77:743-752
35. Muscle Nerve 2015; Online July
36. Neurology 2017; Online March
37. Neurophysiol Clin 2017 Sep 19
38. Nat Genet 2019 Jul 22, Nat Genet 2019 Jul 22, J Med Genet 2021 Oct 21
39. J Neurol 2017;264:1567-1582
40. Continuum (Minneap Minn) 2020;26:116-137
41. GeneReviews
42. Auton Neurosci 2016;197:56-59
43. Sci Rep 2019;9:16982
44. Neurol Genet 2020;6:e496
45.Neurology 2020 Sep 28
46. Acta Neuropathol Commun 2020;8:204
47. J Clin Invest 2021 Jan 26:145837
48. J Clin Invest 2021 Mar 23
49. J Cell Sci 2021 May 20, Hum Mutat 2020;41:1906-1917
50. Endocrinol Diabetes Metab 2021 Aug 10:e00290
51. Front Immunol 2016;6:620
52. Ann Allergy Asthma Immunol 2021 Aug 13:S1081-1206(21)00567-6
53. Ann Allergy Asthma Immunol 2021:S1081-1206(21)01155-8
54. Neurology 2021 Oct 21, J Neurol Neurosurg Psychiatry 2023 Mar 22
55. Clin Genet 2023;104:587-592
56. HGG Adv 2024 Jul 13
57. Skelet Muscle 202;14:15
58. J Pers Med 2024;14:435
59. Alzheimers Dement 2025;21:e14596.
60. Brain 2025 Jun 11
61. Neurol Genet 2025;11:e200261
62. J Pediatr Orthop 2025 Aug 18
63. Nat Genet 2026 Feb 17
2/19/2026