|
Home, Search, Index, Links, Pathology, Molecules, Syndromes, Muscle, NMJ, Nerve, Spinal, Ataxia, Antibody & Biopsy, Patient Info |
Motor Syndromes, Hereditary (SMA, ALS + ...)
|
ALS: Hereditary & Familial Spinal muscular atrophy (SMA): Types Recessive SMA SMA: SMN 5q13 Congenital with arthrogryposis Werdnig-Hoffmann Kugelberg-Welander Spinal muscular atrophy 2 (SMA2) Spinal motor neuropathy: RBM7; 11q232 SMA + Congenital fractures TRIP4; 15q22 ASCC1: 10q22 SMA + Encephalopathy: TBCD; 17q25 SMA + Myoclonus Epilepsy: ASAH1; 8q22 SMA + Pontocerebellar hypoplasia (PCH) PCH1A: VRK1; 14q32 PCH1B: EXOSC3; 9p11 Other: Motor + Cerebellar EXOSC8; 3q13 EXOSC9; 4q27 Mitochondrial DGUOK: 2p13 SCO2: 22q13 SLC25A21: 14q13 TK2: 16q22 Congenital contractures Dominant, Proximal Adult: VAPB; 20q13 Adult + Cramps (SMAJ): CHCHD10; 22q11 Adult: MORC2; 22q12 Bulbar Congenital + Legs weak: TRPV4; 12q24 HMSN-P (Okinawa type): TFG; 3q12 Respiratory & Proximal arms: MAPT; 17q21 Scapuloperoneal syndromes SMALED 1: Leg predominant; DYNC1H1; 14q32 2: Early-onset; Contractures; BICD2; 9q22 X-linked SMA (Recessive) Bulbospinal (Kennedy): AR; Xq12 SMAX 2: Infant + Arthrogryposis; UBE1; Xp11 3: Distal; ATP7A; Xq21 |
HMN D1: UBE3C; 7q36 D2 (2A): HSPB8 (HSP22); 12q24 D3 (2B): HSPB1 (HSP27); 7q11 D4 (2C): HSPB3 (HSPL27); 5q11 D5 (5A): GARS; 7p15 D6 (2D): FBXO38; 5p31 D7 (7A): SLC5A7; 2q12 D8: Congen, Legs: TRPV4; 12q24 D9: WARS1; 14q32 D10: EMILIN-1; 2p23 D11: SPTAN1; 9q34 D12 (5B): REEP1; 2p11 D13 (5C): BSCL2; 11q13 D14 (7B): DCTN1; 2p13 D15; 10q26 R1 (6): IGHMBP2; 11q13 R2: SIGMAR1: 9p13 R3: 11q13 R4: PLEKHG5; 1p36 R5: DNAJB2 (HSJ1); 2q35 R6: REEP1; 2p11 R7: + Myopathy; VWA1; 1p36 R8: SORD; 15q21 R9: COQ7; 16p12 R10: VRK1; 14q32 R11: RTN2; 19q13 + Upper motor neuron Senataxin; 9q34; Dom 4q34: Dom HMN D3: HSPB1; 7q11 D13: BSCL2; 11q13 D14: Dynactin; 2p13 R2: SIGMAR; 9p13 R11: RTN2; 19q13 SPG + Motor neuropathy HMN: 11p; Rec HMN: 16p HMN: KCC3; 15q14; Dom HMN: MME; 3q25; Rec HMN: SLC5A6; 2p23; Rec HMN: KIF21A; 12q12 HMN: BANF1; 11q13 HMN: MAPT; 17q21 HMN/CMT 2KK: ARHGAP19 CMT 2N: AARS; 16q22; Dom CMT 2O: DYNC1H1; 14q32; Dom Neuromyotonia: HINT1; 5q31; Rec Child: BICD2; 9q22; Dom Intellect Disability: TBCK; 4q24; Rec HMN + Tremor: PIGG; 14p6 |
Motor Neuropathy Differential diagnosis General, Distal Lower motor neuron Motor syndromes Protein mechanisms Also: NMJ disorders Distal SMA (DSMA; dHMN) Recessive dHMN: SYT2; 1q32 + Ataxia telangectasia: ATM; 11q22 + Encephalopathy: TBCE; 1q42 Lethal congenital contractures NRCAM: 7q31 Dominant Calf predominant: FBXO38; 5p31 Leg predominant Distal Ulnar-Median Childhood: BICD2; 9q22 + Macular Δ: FBLN5; 14q32 + Hearing loss: MYH14; 19q13 Scapuloperoneal: TRPV4; 12q24 SMAJ1: GARS1; 7p14 HMN: HARS; 5q31 HMN: GBF1; 10q24 HMN: YARS1; 1p35 X-linked dHMN: AIFM1; Xq26 SMAX 3: ATP7A; Xq21 SMARD2: LAS1L; Xq12 Mitochondrial mtATP6 ± Episodic weakness mtATP8 Sporadic: Hirayama Distal Motor Neuropathy or Myopathy |
Multisystem disorders Recessive AAA syndrome: Aladin; 12q13 ANE: RBM28; 7q31 Chediak-Higashi: LYST; 1q42 COMNB: SLC5A6; 22p23 CONDCA: AGTPBP1; 9q21 CONDSIAS: ADPRHL2; 1p34 Hexosaminidase A: HEXA; 15q23 Leukoencephalopathy: SCP2; 1p32 MND + Dementia & Ophthalmoplegia Motor + Cognitive: MCM3AP; 21q22 MPAN: c19orf12; 9q12 NEDCAM: GEMIN5; 5q33 Optic atrophy: c19orf12; 19q12 STAT5B: 17q21 TBX5: 12q24 TTC19: 17p12 Dominant BIBARS Cataracts & Skeletal abnormalities DDPAC: MAPT; 17q21 HMN: EMILIN-1; 2p23 Machado-Joseph: Ataxin-3; 14q32 Myopathy + Paget: HNRNPA2B1; 7p15 X-linked Cabezas: CUL4B; Xq23 Neuroaxonal dystrophy 2 Polyglucosan body: GBE1; 3p12 Mitochondrial: SCO2 Sporadic: Camera-Marugo-Cohen Spastic paraparesis Spastic paraparesis + Motor neuropathy Bulbar syndromes AAA syndrome: Aladin; 12q13; Recessive Brown-Vialetto-van Laere: SLC52A2/A3 BSMA: Androgen Receptor; Xq12 BSMA: Dominant Bulbar ALS Fazio-Londe: Recessive or Dominant FOSMN PLS, Juvenile: Alsin; 2q33; Recessive Worster-Drought |
Distal SMA
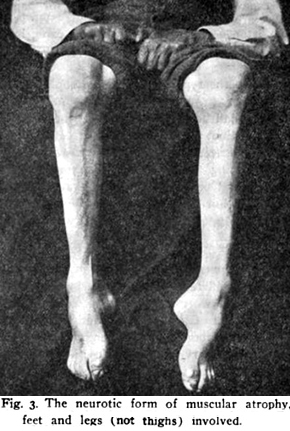 From: Spiller |
HMN: Protein mechanisms
dHMN: Spain 167
- General
- Prevalence: 2.3 per 100,000
- Many sporadic
- Onset age: Often in 1st decade
- Types
Spinal Muscular Atrophy (SMA0; SMA1; SMA2; SMA3; SMA4; SMA 5q)
 178
178
●
Survival Motor Neuron 1 (SMN1)
|
Epidemiology & History Genes Clinical correlations SMN1 SMN2 Modifiers Neighboring & Related SMN Protein Clinical features Congenital Arthrogryoposis SMA 0 Types: 1; 2; 3; 4 Lower motor neuron Pathology Treatments |
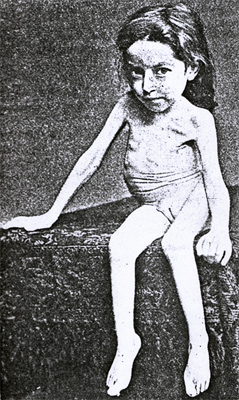 Hoffman ~1891 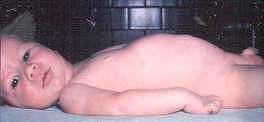 From: Andrew Kornberg MD |
 Guido Werdnig
 Johann Hoffman
|
SMA 5q: Classification (without treatment)
- Inheritance: Recessive
- SMN1 mutations: Bi-Allelic
| SMA Type |
SMN2 Copies |
SMA 5q % |
Onset Age |
Motor Milestone Achieved |
Life Expectancy |
| 0 | 1 | < 1% | Birth | Never Sit | < 6 mo |
| 1 | 2-3 | 55% | 0 to 6 mo | Never Sit | 8 to 24 mo |
| 2 | 2-4 | 30% | 6 to 18 mo | Sit | 2 to 4 decades |
| 3 | 3-5 | 10% |
3A: 1.5 to 3 yrs 3B: 3 to 20 yrs |
Walk | Normal |
| 4 | 3-5 | 5% | Adult | Walk | Normal |
- History
- SMA described independently by Guido Werdnig & Hoffmann in 1891
- Werdnig
- Described condition as "Neurogenic dystrophy"
- Hoffmann
- Established spinal nature of SMA
- Coined term: "Spinale muskelatrophie"
- Epidemiology
- Incidence of SMA disease: 1 in 6,000 to 20,000 births 25
- 2nd most frequent autosomal recessive disease of childhood (After cystic fibrosis)
- Carrier frequency of SMN1 mutations
- General Western population: 1.7% to 2.9%
- Iran: 5% to 5.5% 213
- Sub-Saharan Africa
- Frequency of carriers lower: 0.5%
- SMN1 copy numbers: Higher; 53% with 3 copies vs 6% with 3 in West
- SMN2 copy numbers: Lower; 24% with none vs 8% in West
- SMA 5q: Genetics
SMN1 gene
SMN2 gene
Other
- SMN Gene testing
- SMN deletions in > 95%: Exons 7 & 8 tested for deletions
- Carrier testing
- Quantitation of number of SMN1 genes
- Does not detect carriers with > 1 SMN1 gene on a chromosome
- SMN1 (Telomeric SMN (SMNT)) gene
- SMN1 mutations
172
- Most (95% to 98%)
- Deletion of SMN1 gene: Often deletion of entire gene
- Homozygous in 93%
- Few (4%): Deletion of exon 7; May be de novo
- Rare
- SMN1 point mutation (2%)
- Common: Missense or In-frame deletion
- Also: Splice site & Stop
- Exon 8 deletion
- Gene conversion of SMN1 to SMN2 (Exon 7 C>T)
- Large deletion: SMN1 ± neighboring NAIP gene
- SMN1 point mutation (2%)
- Most (95% to 98%)
- Disease relations
- Number of SMN1 gene copies
8: Varied
- 1 SMN1 copy on each chromosome
- 82% to 96% of normal individuals
- 2 SMN1 copies on one chromosome (Duplication)
- 4% to 18% of normal individuals
- May have deletion on other chromosome
- Variability makes heterozygote testing complicated
- 1 SMN1 copy on each chromosome
- SMN1 gene structure: Strongly homologous to SMN2
- Contains 9 exons
- Exons 7 & 8 contain gene-specific nucleotide sequences
- Some differences from SMN2
- AG-rich exonic splice enhancer in SMN1 exon 7
- Enhancer increases inclusion of exon 7 in protein
- SMN1 gene chromosomal surrounding DNA
- Composition: Large inverted duplication
- Duplication has telomeric (t) & centromeric (c) copies
- Each copy of duplication contains at least 4 genes
- SMN
- Other: p44 (GTF2H2); NAIP; H4F5
- Repeated DNA unit features
- Present on each chromosome
- 0 to 4 copies
- Telomeric (SMN1; SMNT) gene mutations
- Related to presence of SMA disease syndromes
- SMN1 mutations
172
- SMN2 gene (Centromeric SMN (SMNC))
- Gene location: Centromeric to SMN 1 gene
- Strong homology to SMN1: 5 nucleotide differences
- Introns: One difference in 6; Two in 7
- Exons: 2 Differences
- Exon 7 (C to T): Translationally silent
- Exon 8: 3' untranslated region
- Nucleotide differences from SMN1 produce altered SMN splicing
- Can produce identical amino acid sequences to SMN1
- If fully translated
- Quantitative: 10% of SMN1 gene
- Often produces smaller SMN proteins than SMN1
- SMN2 gene transcript often spliced at exon 5 or 7
- 70% to 80% of SMA2 transcripts lack exon 5 and/or exon 7
- Mechanism of altered splicing
- Single-nucleotide change from SMN1 to SMN2
- Location: Heptamer motif of exon splicing enhancer
- New nucleotide sequence (C to T)
- Creates exonic splicing silencer
- Inhibition is hnRNP A1
dependent
37
- Inhibition is hnRNP A1
- ? Eliminates motif recognized by splicing factor
- Heptamer motif recognized by
- Splicing factor, arginine/serine-rich (SF2/ASF)
- Splicing factor, arginine/serine-rich (SF2/ASF)
- Nucleotide change abrogates
- SF2/ASF-dependent exon splice enhancer
- Heptamer motif recognized by
- Creates exonic splicing silencer
- Single-nucleotide change from SMN1 to SMN2
- Can produce identical amino acid sequences to SMN1
- SMN2: Disease relations
202
- Number of Copies: More copies related to less severity of SMA
- SMN2: Gene number
8: One or Multiple copies
- Normals: SMN2 copies
- None 9%
- 1: 42%
- 2: 46%
- 3: 3%
- SMA carriers: SMN2 copies
- None 1%
- 1: 18%
- 2: 47%
- 3: 31%
- 4: 3%
- SMA patients
- 1 or more SMN2 copies
- Reduced SMN2 Gene number correlates with
- Increased SMA severity
- Normals: SMN2 copies
- Homozygous SMN2 Deletions: Disease associations
- More common in Lower motor neuron syndromes & MMN
- Sporadic ALS: No clear effect
- SMN2: Disease-relation modifiers
- c.835–549A>G & c.835–44A>G variants: Milder SMA disease
- c.859G>C substitution: SMA less severe
SMN Genes: Different Splicing
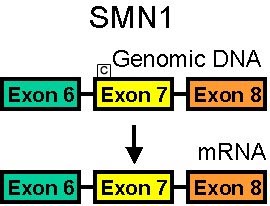
SMN1 mRNA
90% of pre-mRNA spliced to full length SMN
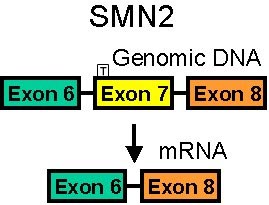
SMN2 mRNA
80% of pre-mRNA spliced to SMN with no exon 7
SMN without exon 7 is unstable & rapidly degraded- SMA modifier genes
- Plastin 3 (PLS3)
- Ca2+-dependent protein
- Overexpression
- Protective in SMA
- Improves endocytosis
- Neurocalcin Delta (NCALD)
- Protein
- Neuronal calcium sensor
- Interacts with: Clathrin
- Endocytosis suppressor
- Suppression
- Mutations: 17-bp deletion upstream of NCALD; SNP rs147264092 in intron 1
- Clinical effect: Protective
- SMN1 mutations with 3 or 4 copies of SMN2
- Patients continue to be photosensitive
- Epidemiology: 2 families
- Protein
- Plastin 3 (PLS3)
- Related or neighboring genes on chromosome 5q
- SMN1 chromosomal locus: General
- Complex 500 kb region of Chromosome 5q13
- Contains large- and small-scale repetitive genetic elements
- Locus of unusual genetic instability
- Frequent spontaneous mutations
- The 500 kb region contains 4 genes: SMN, NAIP, p44, H4F5 (SERF1a)
- The 3 genes are located in a locus of duplicated and inverted DNA (6 genes)
- Telomeric genes are functional genes
- Centromeric genes represent dysfunctional copies
- Genetic structure is unique to humans

- Neuronal Apoptosis Inhibitory Protein (NAIP)
- Females: Absence of NAIP strongly associated with severe phenotype
- Males: No relation between NAIP deletion & phenotype
- GTF2H2 (Btf2-p44)
- Deleted in 15% of SMA patients
- SERF1A (C212/H4F5)
- C212: Multicopy microsatallite marker in an intron of H4F5
- Reduction or absent alleles in
- SMA type I: 94%
- SMA type II: Frequency between type I and controls
- SMA type III: Slightly more frequent than controls
- Closest gene to SMN
- Function unknown
- Retrotransposon-like sequences
- Transcribed into RNA
- Reverse transcribed into cDNA
- Reintegrated as cDNAs into the genome at a new location
- SMN1 chromosomal locus: General
- SMN protein: 38 kD or smaller splice variants
- 294 amino acids
- SMN Expression
- From both SMN1 & SMN2 genes
- Transcript splicing
- SMN1 (SMNT) proteins
- 90% full length
- 10% missing exon 5
- AG-rich exonic splice enhancer in SMN exon 7
- Increases inclusion of exon 7 in protein
- SMN2 (SMNC) proteins
- Full length: 20% to 40%
- Missing exon 5, or 7, or both: 60% to 80%
- Most SMN2 spliced to generate short isoform of SMN protein without exon 7
- Less stable: Short half-life
- Poor self-oligomerization
- SMN1 (SMNT) proteins
- Anatomical locations
- High levels: Brain & spinal cord; Kidney; Liver
- Moderate levels: Skeletal & cardiac muscle
- Cell locations: Nuclear & cytoplasmic inclusions (gems)
- Nucleus: 34kDa variant
- Localized to gem bodies & nucleoli
- Near nuclear membrane
- Cytoplasm: 38kDa variant
- Especially in motor neurons
- Sub-cellular locations
- Perikarya; Proximal dendrites; Axons
- Motor neurons: May be associated with cytoskeletal elements & mitochondrial membranes
- Most cells: Not associated with organelles
- Nucleus: 34kDa variant
- Levels in tissue
- Highest during development
- Downregulation of SMN: From early postnatal periods to adulthood
- SMN Interacts with other proteins
- General
- Self oligomerization: Via N- & C-terminus (Exon 7)
- SMN & Gemins 2 to 5 form a multiprotein complex
- Assembles in the cytoplasm
- Translocates to nucleus: Located in gems
- Complex function: Cytoplasmic formation, nucleus import, & regeneration of spliceosomal snRNPs
- SIP1 (Gemin 2)
- Strong interaction: forms heteromeric complex with SMN
- Attachment Via N-terminus of SMN
- Location: Gem bodies in nucleus
- Localization
- Colocalizes with SMN in nucleus & cytoplasm
- SMN does not colocalize with SIP1 in neurites of motor neurons
- SMN & SIP1 form part of protein complex with Sm core proteins & snRNP U1
- Other Gemins
- Gemin 3 (dp103)
: DEAD box putative RNA helicase
- Gemin 4

- Gemin 5
- Gemin 6
- Gemin 3 (dp103)
- Spliceosomal snRNP core proteins (Sm)
- B/B'; D1-3; E; F; G
- Via central tudor domain
- Needed for U snRNP assembly: 5'cap hypermethylation; Import into nucleus
- Spliceosomal snRNA U1 & U5
- Via N-terminus of SMN
- Located in cytoplasm
- HnRNP protein U
& fibrillarin
:
Located in nucleoli
- Bcl-2
:
Coexpression with SMN
- Provide synergistic protection against Bax
or Fas
induced apoptosis
- Provide synergistic protection against Bax
- Nuclear transcription activator E2 of papillomavirus: Via C-terminus
- Profilin 2
- Motoneuron-specific microfilament-associated, actin-binding protein
- Action: Inhibits polymerization of actin
- SMN interaction: Via Pro5-X17-Pro10-X17-Pro5 motif encoded by exons 4, 5 and 6 of SMN gene
- General
- SMN: General functions
51
- Nuclear: Regeneration of active splicing complex
- Part of complex regulating assembly of spliceosomal U snRNPs (RNA-protein complex)
- Essential component of the spliceosome that catalyses pre-mRNA splicing
- Linked to control of protein synthesis & to expression of new protein isoforms
- Axons
- May play role in axonal transport of mRNA
- Promotes neurite outgrowth and/or neuromuscular maturation
23:
Downregulation causes
- Aberrant guidance of axons with excessive axonal branching
- Reduced growth velocity
- Reduced growth cone sizes
- Anomalous calcium-channel clustering in growth cone
- Abnormal presynaptic motor axon nerve terminals
- Presynaptic defects in synaptic vesicles, mitochondria, active zones, neurofilaments & microtubules
- Axonogenesis: May be related to spliced isoform of SMN protein
- Presynaptic motor axon terminals: Postnatal organization & maintenance 84
- Muscle: SMN knockout produces myopathy
- Disease correlations
- SMN mutant proteins: Reduced RNA-binding activity
- Less SMN protein & gem bodies in more severe SMA types
- Mutations: Impaired endocytosis
- Nuclear: Regeneration of active splicing complex
- SMN: Neuromuscular localization
- Nerve
- SMN accumulates in growth-cone-like structures during neuronal differentiation
- Selective deletion of SMN in nerve causes loss of motor neurons
- Muscle
- SMN concentrated at NMJs
- Cytoplasmic SMN high during NMJ formation & reduced with NMJ maturation
- Selective deletion of SMN in muscle produces myopathy 24
- Nerve
- Splicing functions of SMN: Ubiquitous
- Interacts with both RNA-binding proteins and RNA
- RNA-binding element in exon 2a
- Post-transcriptional nuclear RNA metabolism
- During spliceosomal snRNP biogenesis and pre-mRNA splicing
- Related to spliceosomes
- Spliceosomal snRNPs: SMN-related proteins promote transport from cytoplasm to nucleus
- Inhibition leads to
- Reduced spliceosomal snRNP biogenesis
- ? Reduced conversion of pre-mRNA (with introns) to mRNA (no introns)
- SMN Autoantibodies: Clinical associations
199
- Systemic sclerosis + Myositis
- Calcinosis
- Trigeminal neuropathy
- GI involvement, Lower
- Severe phenotype
- Anti-U1RNP + Mixed Connective Tissue Disease (MCTD)
- Skin: Fingertip pitting scars
- Myopathy
- Myocarditis
- Gastrointestinal, lower
- Systemic sclerosis + Myositis
- SMN: Clinical - Genetic correlations 2
- SMN1 (SMNT; telomeric SMN gene) mutations & disease
- SMN1 mutations present in 95% of SMA patients
- SMN1 & SMN2 genes: Correlation with disease severity
- Milder disease (SMA II or III)
- Increased SMN2 gene copy number
- Absence of both SMN1 & SMN2 genes
- Lethal
- SMA type 0: No SMN1 gene & 1 copy of SMN2
- Severe weakness
- Death < 1 month of age
- SMA type I: Mutations
- Mostly SMN1 deletions
- Few missense point mutations in SMN1
- SMN2 gene copy number: Often 2
- SMA type II
- Mutations convert SMN1 gene to SMN2
- SMN2 gene copy number: > 3
- Missense point mutations more common
- SMA type III
- SMN2 gene copy number: > 3
- Missense point mutations more common
- Milder disease (SMA II or III)
- Total amount of full length SMN protein
- ? Best correlation with SMA severity
5q CHROMOSOMES
Typical SMN mutations in SMA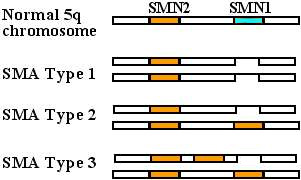
SMN1 Normal gene 
SMN1 Mutation types
Deletion :
More severe SMA
:
More severe SMA
Conversion to SMN2 gene :
Milder SMA
:
Milder SMA
SMN2 gene: Variations
More copies: Correlate with milder SMA.
SMN2 mutations alone: Don't produce SMA- SMN1 (SMNT) deletions
- Usually involve exons 3, 6, 7 & 8
- 95% of SMA: Exon 7 of SMN1 gene homozygous absent
- Rare homozygotes for selective 7 & 8 deletion
- Clinically normal, or only mildly weak
- Larger SMN deletions
- Associated with earlier onset & more severe disease
- May affect other neighboring genes
- Hybrids of SMN2 (centromeric)
& SMN1 (telomeric)
SMN genes may occur
- Centromeric exon 7 & telomeric exon 8
- Usually associated with SMA 2 or 3
- Mechanisms producing SMN1 deletions in offspring
25
- Both parents with one SMN1 copy
- SMN1 mutations on one chromosome in each parent
- Recurrence risk for another child with SMA: High (25%)
- De novo rearrangements
- New SMN1 deletion in gene from 1 parent: Parental SMN1 copy number is 2
- Other parent is carrier of SMN1 deletion: SMN1 copy number is 1
- Frequency: 2% of SMA
- Mechanisms of rearrangements: 2 Diferent types
- Unequal crossing over between repeated units during paternal meiosis
- Conversion of part of SMN1 gene into SMN2: Exon 7 of SMN2 & Exon 8 of SMN1
- Recurrence risk for another child with SMA: Low
- SMN duplication in cis: One parent with 2 SMN genes on one allele and 0 on other
- Normal total SMN1 copy number: 2
- 50% chance of transmitting gene with SMN1 deletion
- Recurrence risk for another child with SMA: High (25%)
- Frequency of SMN duplication in cis in SMA parents: 3% to 8%
- Frequency of SMN duplication in cis: 0.1% of population
- Both parents with one SMN1 copy
- Deletions in SMN2 gene alone not related to SMA
- SMN1 (SMNT) missense point mutations
- Frequency: Rare
- 3.6% of 5q13 SMA
- Highest in type III SMA
- Lowest in type I SMA
- ? Higher in some non-European populations: South African blacks
- Other allele: SMN1 deletion
- Location
- Cluster at the 3' end of gene
- Exon 6: Between codons 258 to 279
- Modular oligomerization domain
- Correlation between residual SMN oligomerization (self-association) & severity
- SMN I: Severe loss of function mutations (G279V & Y272C)
- SMN II & III: T274I & S262I
- In tyrosine/glycine-rich motif also present in RNA binding proteins
- Exon 3 termination mutations (425del5; W102X): Exon skipping; Mild phenotype
- Mutation consequences
- Most interfere with SMN oligomerization
- E134K: Alters binding of Sm proteins; Severe SMA phenotype
- Clinical: SMA II & III phenotypes common
- Frequency: Rare
- SMN2 (SMNC)
- Copy number associated with SMA disease severity
- c.859G>C substitution (G287R) 69
- Epidemiology: 3 patients described
- Genetics
- SMA type III patients
- Have fewer SMN2 copies than expected
- Have copies of mutated (G287R) SMN2 gene alone, or in combination with usual SMN2 gene
- Normal population: Mutated SMN2 not common
- SMA type III patients
- SMN protein
- More full length SMN protein produced
- Mutation effect related to splicing modification
- Clinical: SMA less severe than expected from SMN2 copy number
- Proximal strength: Mildly to moderately reduced; Worst in hip flexors & knee extensors
- Patients often SMA type III
- SMN2 Deletions
20: Homozygous
- Genetics
- Mutation: Deletion of exon 7 in SMN2 gene
- Normal population: Homozygous deletion in 5% to 9%
- Increased incidence of homozygous SMN2 deletion in D-LMN syndromes: 36%
- Clinical: Distal lower motor neuron syndromes
- Weakness: Distal; Asymmetric; Upper & Lower extremities
- Course: Rapidly or slowly progressive
- Differences from other LMN syndromes
- Earlier age of onset (40 vs 56 years): Some as young as 15 years
- Lower preponderance of males: M:F ratio of 1.5 vs 2.5
- Both arms & legs involved
- Genetics
- SMN mutations not found in sporadic or familial ALS
- Neuronal Apoptosis Inhibitory Protein (NAIP)
deletions
- Occur in 35% of SMA patients
- In SMA: Virtually always occur with SMN deletion
- Involve exon 5
- Rarely also occur in unaffected
More common in SMA type I (45% - 66%) than in II or III (5% - 16%)- NAIP absence: Correlates with severe phenotype in females, not males
- Rare (2%) SMA 5q patients with neither SMN or NAIP deletion
- Modifier protein: Plastin 3 (PLS3)
- Unaffected SMN1-deleted females: Higher expression
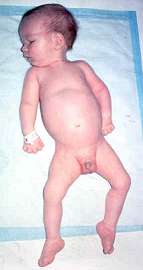
- Clinical features: Congenital SMA (5q) with arthrogryposis
- Onset age: Congenital
- Severe hypotonia
- Movements: Absent; Respiratory failure at birth
- Cranial nerves: Facial diplegia; ± External ophthalmoplegia
- Contractures: Especially knees
- Course: Death < 30 days
- Pathology
- Loss of motor & sensory myelinated axons
- Motor neurons: Preserved, swollen
- Rule out X-linked SMA
- Clinical features: SMA Type 0
138
- Genetics
- Onset age: Prenatal
- Fetal
- Movements: Reduced
- Nuchal translucency
- Gestational age at delivery: Mean 39 weeks
- Muscle weakness: Severe
- Hypotonia: Severe
- Few spontaneous movements
- Respiratory failure
- Cranial nerves: Inability to suck or swallow; Face
- Posture: Frog-like
- Tendon reflexes: Absent
- Tongue fasciculations
- Fetal akinesia deformation sequence
- Contractures: At one or multiple joints
- Micrognathia
- Palate: High arched
- Edema, peripheral
- Intrauterine growth retardation (50%)
- Cardiac
- Congenital defects (85%): Septal
- Bradycardia
- Extraocular movements: Normal
- Course
- Untreated: Death in 1st month
- Treatment
182
- Prolonged survival
- Motor
- Respiratory insufficiency
- GI: G-tube dependent for nutrition
- Weakness: Severe; Some anti-gravity movement
- Skin: Rash; Necrosis & Vasculopathy
- Cardiac: Malformation
- Bones: Fractures
- Clinical features: Werdnig-Hoffmann (Type 1)
- Onset
- Usually before 3 months
- Range
- 0 to 6 months
- Some with in utero decreased fetal movements
- Acute onset in occasional patient
13
- Early weeks or months: Normal development
- After 1st few months
- New weakness
- No new motor milestones
- Motor neuron loss
- Clinical
- Weakness
- Diffuse; Proximal > Distal
- Severe
- Poor feeding
- Respiratory insufficiency: Paradoxical respirations
- Sparing of facial & oculomotor
- Hypotonia
- Fasciculations: Tongue
- Tendon reflexes: Reduced or absent
- Scoliosis
- Intellect: Normal; Alert faces
- Prognosis
- Untreated
- Respiratory failure
- Death
- 50% by 7 months
- 95% by 17 months
- Chronic course in 5%
- Treatment
- Better prognosis with: Earlier treatment
- Survival: Prolonged; 100% at 20 months
- Ventilator free: Often
- Motor: Improved strength & Milestones
- Feeding: Oral
- Hospitalizations: Reduced
- Untreated
- Weakness
- Pathology
- Muscle
- Large regions of grouped muscle fiber atrophy
- Most larger fibers are type I
- Spinal cord
- Anterior horn: Motor neuron loss
- Cell pathology
- # of gem bodies in fibroblasts correlates with disease severity
- SMA I spinal cords: 100-fold reduction in SMN protein
- Muscle
- Variant: Facial weakness & Ophthalmoplegia
From: A Kornberg MD - Onset
- Clinical features: Kugelberg-Welander (Types II
& III
)
- Classification
- Type II: Intermediate
- Onset: Often < 18 months
- Never stand
- Life span
- Often shortened
- Death > 2 years
- Rule out: SMA2
- Type III: Mild
- Onset: Often > 18 months
- Stand independently
- Life span
- ± Shortened
- Death in adulthood
- Type II: Intermediate
- Clinical
- Onset
- Childhood or Juvenile
- Cramps may be 1st symptom
- EMG: Mild neurogenic changes
- Weakness
- Proximal
- Symmetric
- Posterior > Anterior: Triceps; Gluteus; Hamstrings
- Variable degrees of severity
- Some never walk
- Poor prognosis
- Scoliosis early
- Later onset: Better prognosis
- Respiratory
- Sleep disorders with vital capacity < 65%
- Some never walk
- Progression
- Most have loss of function over time
- ? Change in strength over time
- Difficult to measure
- Electrophysiology
- Progressive loss of motor units
- Loss of ambulation
- Tremor
- Tendon reflexes: Reduced
- Fasciculations
- Cognition: Normal 169
- Metabolic syndrome 208
- Fertility disorders: Spermatogenesis 214
- Onset
From: M Ryan MD- Clinical features: SMA 5q - Later onset (Type IV)
- Onset ages: Juvenile or Adult
- Weakness
- Distribution
- Proximal
- Patchy: Psoas; Quadriceps > Hamstrings; Triceps
- Symmetric or Asymmetric
- Progression
- Most: Very slow loss of function over time
- ? Change in strength over time: Difficult to measure
- Distribution
- Fasciculations
- Tendon reflexes: Reduced
- Course
- Often continue ambulatory
- Lifespan: Normal
- Hydrocephalus
173
- 4x increased frequency
- May be obstructive
- Lower motor neuron syndromes
- Genetics
- SMN2 deletions
- Increased incidence of homozygosity
- Distal lower motor neuron syndromes
- 36% vs 5% in general population
- Occasional homozygous SMN1 deletions
- SMN2 deletions
- Clinical
- Distal > Proximal
- Asymmetric
- Progressive weakness
- Slower with SMNC than with SMNT deletion
- No family history
- Muscle MRI: SMA II & III 218
- More involvement
- Psoas major, Soleus, Rectus femoris
- Less involvment
- Biceps brachii, Deltoid, Pterygoid medial
- Other
- Tongue (50%): Associated with dysphagia
- Thigh: Anterior > Posterior; Rectus femoris, Quadriceps, Vasti
- Pathology
- Muscle Pathology
- Grouped atrophy
- Large fibers: Mostly type 1; Hypertrophied
- Small fibers mixed or type 2
- Development
- Weakness & Muscle atrophy: May precede motor neuron loss
- Spinal cord
26
- Motor neurons in anterior horn
- Loss
- Reduced by 50% to 70% in type 1 SMA
- Time period
- 12 weeks of gestation to birth
- Similar to period of normal cell death
- Apoptosis
- Increased: Only at 12 to 16 weeks, Not post-natal
- Remaining motor neurons
- Size: Small
- Chromatolytic changes
- Anterior roots: Atrophic
- Loss
- Clarke’s column sensory–associated neurons: Injury features
- Sympathetic lateral horn: Normal
- Post-natal
- Morphological changes in some motor neurons
- Reduction in central Nissel
- Peripheral displacemnet of nuclei
- Spinal cord size: 40% reduced
- Descending corticospinal tract: Larger
- Morphological changes in some motor neurons
- Motor neurons in anterior horn
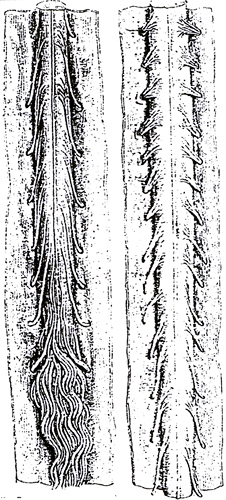
SMA Spinal cord
Anterior roots are atrophic- SMA 5q: Treatments 164
- SMN2 pre-mRNA splicing
- Modify: Increase full length SMN protein
- Drugs
- Nusinursen (Spinraza)
- Drug type: Anti-sense oligonucleotide
- Mechanism
- Binds to splice-silencing-site in SMN2 intron 7
- Inhibits action of other splice-factors
- Treatment route: Intrathecal; Repeated treatments
- Effects: Improved strength; Prolonged survival
- Most effect: Youngest patients
- Serum creatinine to cystatin C ratio
- Mild increase 222
- Risdiplam
- Drug type: Small molecule
- Mechanism
- Binds 2 sites on SMN2 pre-mRNA
- Full-length SMN mRNA & protein: Levels ↑
- Treatment route: Oral
- Benefit: Improved survival & strength
- Nusinursen (Spinraza)
- SMN1 gene delivery: Onasemnogene abeparvovec (Zolgensma)
- Non-replicating adeno-associated virus capsid (scAAV9)
- Delivers wild-type SMN1 gene to motor neurons
- Drug route: Intravenous; single treatment
- Effects
- Most effect: Youngest patients
- Improved strength
- Prolonged survival
- SMN2 copies: Better with 3 than 2
- CMAP amplitudes: Increased or Unchanged
- SMA1: More satisfactory responses than nusinersen 225
- Non-replicating adeno-associated virus capsid (scAAV9)
- Experimental treatments
- Branaplam: SMN2 splicing
- Reldesemtiv (Tirasemtiv): Fast skeletal troponin inhibitors
- Myostatin inhibitors: SRK 105 (Monoclonal antibody)
- Olesoxime: Anti-apoptotic agent
- Hydroxy-Urea: No benefit in SMA2 & SMA3
- Histone deacetylase (HDAC) inhibitors: Non-specific increase
- Phenylbutyrate
- Valproate: 250 mg to 500 mg bid po
- Possible mechanism: Increase SMN2 expression
- Care & Treatment
- Respiratory support
- Scoliosis care
- Nutrition
- Mouse models
- Mice normally have no SMN2 gene
- Absent SMN1 with transgenic SMN2 expression
- More SMN2 expression → Less severe disease
- SMN1 heterozygotes with ~50% reduction of SMN protein
- SMA Type 3 phenotype
Spinal Muscular Atrophy: Treatments Features Drug Nusinersen Risdiplam Onasemnogene
Abeparvovec-xioDrug Type Oligonucleotide,
AntisenseSmall Virus (AAV)
Gene DeliveryMechanism More splicing of SMN2 gene to
full length SMN proteinSMN transgene: Produces
full length SMN proteinFDA: Ages All > 2 months < 2 years Pre-Rx Platelets
Coagulation
UrinalysisLiver functions
Platelets
Troponin-I
AAV9 antibodies
Prednisolone Rx: 30 daysRx Intrathecal
q 2 weeks x4,
then q 4 monthsOral
DailyIntravenous
Single doseLimitations Lumbar puncture
neededDrug
interactionsPre-Rx serum
AAV9 antibodiesAdverse
EventsThrombocytopenia
Proteinuria
LP problemsFever
Diarrhea
Skin rashLiver Δ
Acute; Transaminitis
Torticollis
Thrombocytopenia
Troponin ↑Monitor Platelets
Coagulation
UrinalysisLiver functions
Platelets
Troponin-I - Classification
Bulbo-Spinal Muscular Atrophy (SMAX1; BSMA; SBMA; Kennedy Syndrome)
●
Androgen Receptor (AR) (Increased CAG repeats)
|
Androgen receptor protein Clinical features Clinical-genetic correlations Epidemiology Laboratory features Onset Pathogenic mechanisms Pathology |
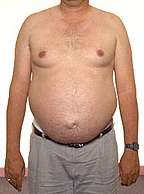
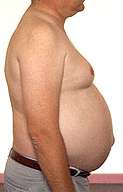 Bulbo-Spinal Muscular Atrophy
Gynecomastia |
||
|
|
- History: 1st description
- Hiroshi Kawahara: 1897
- 2 brothers and an uncle with tongue fasciculations & progressive weakness
- Epidemiology
- Most common adult onset SMA
- General frequency: 1 in 6,887 to 50,000 192
- SBMA especially common in
- Vasa region of western Finland: Scandanavian founder haplotype
- Some regions in Japan: Founder effect
- Western Canada (Indigenous ancestry)
- Other founder effects
- Different founder haplotypes in various European & Asian countries
- No founder haplotype identified in Canadian patients
- Genetics
- Mutation: Increased CAG repeats
- Allelic disorders
- Females
- Androgen insensitivity, Recessive
- Androgen insensitivity, partial ± Breast cancer, Recessive
- Hypospadias 1, X-linked Recessive
- Spinal and bulbar muscular atrophy of Kennedy
- Prostate cancer, susceptibility to, Somatic, Dominant
- Genetic-Clinical Correlations: CAG repeat length
- Normal CAG repeats
46
- Length: 9 to 34 repeats
- Median lengths in populations
- African American: 19–20
- White Caucasian: 21–22
- Asian: 22–23
- Hispanic: 23
- Location: Exon 1
- Intermediate CAG repeats
- Length: 35 to 39
- May manifest with later disease onset age
- BSMA: Long CAG repeats
- BSMA: 40-68 CAG repeats
- CAG repeat length effects
- Longer
- Earlier disease onset
- ? More severe SBMA disease
- Impaired spermatogenesis
- More somatic mosaicism
- No effect on specific clinical features
- Length inversely correlated with transcriptional activity by the androgen receptor
- Longer
- Transmission
- Normal repeat lengths: Transmitted without change; De novo mutations rare
- Expanded repeats: Expansions or contractions in 25% of meiotic events
- Paternal transmission: Larger expansions in CAG repeat number
- Maternal transmission: Contractions or expansions in CAG repeat number
- Prostate cancer
- Fewer CAG repeats (< 18)
- Increased Frequency, especially at young age
- Extraprostatic extension, distant metastases, or high histologic grade
- Prostatic tumor cells
- Dual somatic missense mutations within the AR-CAG repeat
- (CAG)22CAA to (CAG)12CTG(CAG)6CTGCAA
- Interrupts PolyQ tract with 2 leucines
- Dual somatic missense mutations within the AR-CAG repeat
- AR point mutations: Metastatic prostate cancer cells
- Fewer CAG repeats (< 18)
- Androgen insensitivity
- Complete
:
Premature stop codons
- Partial
- Reifenstein Syndrome
:
Point mutations
- Kennedy's syndrome: Bulbo-Spinal Muscular Atrophy
- Reifenstein Syndrome
- Complete
- Other disorders associated with AR-CAG repeat length
- Hereditary hearing impairment
- Schizophrenia
- Benign prostatic hyperplasia
- Risk of developing breast & endometrial cancers
- Normal CAG repeats
46
- Androgen receptor (AR) protein
- Family: Steroid/thyroid hormone receptor
- Phosphoprotein
- Domains
- N-terminal: Contains polyglutamine repeat (Exon 1)
- Central
- DNA binding
- Contains 9 invariant cysteine residues
- Binds zinc ions in 2 zinc fingers
- Nuclear localization signal
- C-terminal
- Binds 2 biologically active ligands
- Testosterone
- Dihydrotestosterone
- Binds 2 biologically active ligands
- Cell locations
- Unbound receptor in aporeceptor complex with heat shock proteins
- Hormone binding
- Receptor translocates to nucleus
- Binds as dimer to DNA through zinc finger domain
- Neuronal function: Plays role in cell survival and dendritic growth
- External link: Androgen Receptor
- BSMA pathogenic mechanisms
- General: Toxic gain of function → Neuronal degeneration
- Large polyglutamine tract: Molecular effect
- Partial proteolysis due to abnormal folding of Androgen receptor
- Truncated forms of androgen receptor may be produced
- Aggregate location: Cytoplasm & Nucleus
- Aggregates
- May contain
- Androgen receptor fragments: Only N-terminal epitopes
- Proteasome components & ubiquitin; PA700 proteasome caps
- Chaperones & Nuclear components with long polyglutamine tracts
- Mitochondria
- Other: Steroid receptor coactivator 1; NEDD8, Hsp70, Hsp90;
HDJ-2/HSDJ; CREB binding protein
- Formation suppressed by HDJ-2 chaperone
- ? Toxic to cell
- Nuclear location of aggregates may be necessary for toxicity
- Truncated forms of AR with expanded repeat
- More toxic than full length protein
- No correlation between frequency of aggregates & cytotoxicity
- May contain
- Proteins possibly involved in mechanism of CAG repeat toxicity
- Caspases: May cleave androgen receptor
- CREB binding protein (CBP)
42
- Polyglutamine-expanded androgen receptor
- Interferes with CBP-mediated transcription of VEGF
- VEGF: May rescue cultured cells with mutant androgen receptor
- Polyglutamine-expanded androgen receptor
- Sir2
pathway
50
- Increased Sir2.1 levels reduce polyglutamine repeat toxicity
- Resveratrol may reduce polyglutamine repeat toxicity
- Loss of Androgen Receptor function
- Non-CAG repeat mutations in androgen receptor reducing function
- Do not produce BSMA by themselves
- Reduced androgen receptor function with CAG expansions
- May exacerbate BSMA disease
- Non-CAG repeat mutations in androgen receptor reducing function
- Clinical features
- Onset
- Age: Mean 27 to 43 years; Range 14 to 75 years
- Early symptoms & signs: Adolescence
32
- Muscle discomfort: Cramps or Pain
- Fatigue: General; Chewing
- Gynecomastia: May be asymmetric
- Weakness: Not common early; May be distal
- Symptoms at 30 years
- Lower > Upper limb weakness
- Occasionally cramps
- Weakness
- Distribution
- Proximal
- Symmetric or Asymmetric (55%; Dominant side 70%)
- Legs > Arms
- Face: Upper & Lower
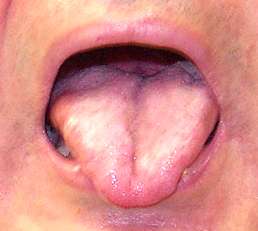
- Tongue: Weakness; Atrophy; Fasciculations
- Proximal
- Bulbar dysfunction
- Dysphagia: Aspiration
- Dysarthria
- Masseter weakness
- Symmetric
- Cold temperatures: Increased weakness
- Slowly progressive: Over decades
- Distribution
- Other muscle features
- Fasciculation-like movements
- Fasciculations
- Atrophy: Especially face & tongue
- Cramps: 50%
- Tendon reflexes: Absent or reduced
- Sensory: Often subclinical changes
- Vibration: Reduced; Legs > Arms
- Sensory Nerve Action Potentials (SNAPs): Small amplitude
- Tremor
- Hands
- Postural & Action
- Early disease manifestation: 4th decade
- NO upper motor neuron signs
- Systemic
- Nonalcoholic fatty liver disease: Most patients detected by MRS
- Androgen insensitivity related
- Gynecomastia (50% to 70%)
- Reduced fertility: Oligospermia
- Testicular atrophy
- Erectile dysfunction
- Groin hernia: 33%
- Other endocrine
- Diabetes mellitus in some patients: Fasting glucose high in 40%
- Pituitary microadenoma: Rare
- Onset
- Laboratory
- Electrodiagnostic
- NCV
- CMAP amplitude: Reduced in 23% to 40%
- SNAP amplitude: Reduced in 80% in sural
- Distal latencies: Prolonged with cold exposure
- EMG
- Face Twitching: Not typical fasciculations
- Individual discharges: Large amplitude; Asymmetric; Repetitive (Grouped)
- Frequency of discharges: Lower in BSMA (~3/min) than in ALS (~20/min)
- Locations: Face (Especially lower), Tongue, Trunk & Limbs
- Onset: Contraction evoked or Spontaneous
- Electrodiagnostic: Grouped axon discharges (Multiple motor units in each discharge)
- Not: Combined fasciculations, Myokymia or Neuromyotonia
- Fasciculations
- Location: Limbs
- Pattern: Single motor units, Large amplitude potentials
- Neuropathy: Chronic partial denervation with reinnervation
- Fibrillations: Legs > Arms; Less common in arms than in ALS
- Motor units: Large, Long, Polyphasic
- Recruitment: Reduced
- Insertion activity: Increased
- Face Twitching: Not typical fasciculations
- Cortical SSEPs: Absent or Prolonged latencies
- NCV
- Serum
- CK: High (94%); May be elevated early in disease course
- Serum estradiol & gonadotropin: Elevated
- Lipid disorders
- Type II hyperlipoproteinemia
- Type IV hyperlipoproteinemia
- Hypobetalipoproteinemia
- Abnormal
- Total cholesterol (54.7%)
- Low-density lipoproteins cholesterol (40%)
- Triglyceride (48%)
- Muscle MRI
207
- Early involvement: Posterior calf
- Other with disease progression: Thigh, Trunk, Anterior calf, Upper extremities
- Muscle biopsy
- Chronic partial denervation
- Grouped atrophy of muscle fibers
- Small angular muscle fibers: Occasional
- Muscle fiber hypertrophy
- NO fiber type grouping
- Internal nuclei
- Type I muscle fiber predominance
- CNS pathology
- Lower motor neurons
- Reduced number in spinal cord & brainstem
- More loss of small motor neurons than ALS
- Sensory ganglion cells: Reduced number in dorsal root ganglia
- Aggregates
- In motor neurons, skin & other tissues expressing androgen receptor
- Intranuclear & Cytoplasmic
- Lower motor neurons
- Electrodiagnostic
- Variant: Patient with 68 CAG repeats
115
- Clinical
- Onset age: 18 years
- Fatigue
- Weakness: Face; Tongue; Proximal; Toes
- Fasciculations
- Cramps
- Tremor
- Dysesthesias: Distal legs & Hands
- Sensory loss: Distal; Panmodal
- Gynecomastia
- Laboratory
- Autonomic: Reduced sweating; Postural tachycardia
- Skin axons: Normal
- Clinical
- Clinical manifestations: Females
- Heterozygous
- General: Mild manifestations later in life
- Muscle cramps: 58%
- Fasciculations: 20%
- Tongue atrophy & fasciculations: 20% in 7th & 8th decade
- Electrophysiology: Chronic denervation in 57%
- Homozygous females
206
- Muscle: Cramps & Twitches in limbs & Trunk
- Hand tremor
- EMG: Mild denervation in 1 patient
- Heterozygous
- Eternal link: e-Medicine
Bulbo-Spinal Muscular Atrophy with Gynecomastia (Autosomal Dominant) 1
● Autosomal Dominant- Onset
- 2nd or 3rd decade
- Nasal voice
- Postural tremor
- Lower motor neuron syndrome
- Tongue: Atrophy; Fasciculations
- Limbs
- Weakness: Mild asymmetry; Proximal + Tibialis anterior
- Atrophy
- Fasciculations
- Tendon reflexes: absent
- Normal: Sensation & Autonomic function
- Cranial nerves: Decreased Upgaze & Convergence (50%)
- Systemic
- Gynecomastia
- No other signs of androgen insensitivity
- Laboratory
- Serum CK: Mildly elevated
- EMG: Acute & Chronic denervation
- Motor evoked potentials (Magnetic stimulation: Prolonged central conduction time
Spinal Motor Neuropathy 150
● RNA-binding motif protein 7 (RBM7)- Epidemiology: 1 Palestinian patient
- Genetics
- Mutation: Homozygous; c.236C>G (p.Pro79Arg)
- RBM7 protein
- Clinical
- Onset
- Age: 1 month
- Hypotonia
- General
- Size (Height, Head circumference & Weight): Low
- Weakness
- Diffuse
- Respiratory
- Muscle: Atrophy; No fasciculations
- Tendon reflexes: Reduced
- Death: 28 months
- Onset
- Laboratory
- Muscle: Grouped fiber atrophy; Large fibers all type 1
- Brain MRI: Normal
Spinal Muscular Atrophy 2 5
● Autosomal Recessive (Not linked to SMA 5q)- Clinical
- Onset: 1 year
- Weakness
- Proximal > Distal
- Symmetric
- Legs > Arms
- Motor functions: Sit but never stand or walk
- Hypotonia
- Muscle atrophy
- Tendon reflexes: Absent
- Tremor: Mild
- Skeletal: Small head circumference; Pes valgus
- Cognitive: Normal
- Progression: ?; May develop respiratory disorders
- Laboratory
- Serum CK: Normal
- Electrophysiology
- EMG: Fibrillations; Large amplitude action potentials
- NCV: Small amplitude CMAPs; Mild slowing; Sensory normal
- Muscle biopsy
- Grouped atrophy
- Type I muscle fiber predominance
Motor Neuropathies: Hereditary (dHMN & HMN) 49, 85, 152
Hereditary Motor Neuropathies: General features
|
|
Distal hereditary motor neuropathy I (HMND1; Distal HMN I)
59
●
Ubiquitin Protein Ligase E3C (UBE3C)
- Nosology
- Juvenile ALS
- Distal HMN
- Epidemiology: Australian family
- Genetics
- Mutation
- 1.35 Mb complex structural variation
- Intrachromosomal translocation
- Transcript (UBE3C-IF): Lacks 13 of 23 exons of wildtype UBE3C
- Effect: Dominant negative
- Inserted sequence fragment contains
- 4 protein-coding genes & their regulatory elements
- MNX1(HB9)
- NOM1
- RNF32
- LMBR1
- Upstream regulatory elements & first 10 exons of ubiquitin-protein E3 ligase gene E3C
- 4 protein-coding genes & their regulatory elements
- 1.35 Mb complex structural variation
- Allelic disorder: Angelman-like syndrome, Recessive 191
- Mutation
- UBE3C protein
- Ubiquitin E3 protein ligase
- HECT (homologous to E6-AP carboxyl 13 terminus) class of E3 ligases
- Forms thioester complex with ubiquitin in presence of an E1 enzyme & E2 enzyme UBE2D1
- Mutation causes: Duplicated copy of 1st 10 exons
- Clinical
- Onset
- Age: Median 10 years; Range 3–40 years
- Disordered walking & running
- Weakness
- Legs: Ankle extensors & Intrinsic foot muscles
- Hands: Mild or None
- Symmetric
- Atrophy: In regions of weakness
- Upper motor neuron
- Muscle tone: Increased (67%)
- Extensor plantar response (56%)
- Tendon reflexes: Preserved
- Sensory: Normal or Vibration reduced
- Foot deformities: Pes cavus; Hammer toes
- Progression: Slow
- Onset
- Laboratory
- Electrophysiological studies
- Motor NCV: Median normal; Tibial slowed; CMAPs normal
- SNAPs: Reduced with age
- Electrophysiological studies
- Nerve pathology: Axonal loss
- MRI: Normal
Distal hereditary motor neuropathy, type 2 (HMND2; HMN 2A; Distal HMN II)
221
●
Heat-shock 22-kD protein 8 (HSPB8; HSP22)
- Epidemiology: 5 families
- Genetics
- HSPB8 mutations: Missense; Lys141Asn, Lys141Glu
- Allelic disorders
- HSPB8 protein
- Heat shock protein
- Chaperone
- ATP-independent holdase
- Prevents aggregation of misfolded proteins
- Participates in refolding process
- Interacts with ATP-dependent foldases: Heat shock protein 70
- High expression in motor & sensory neurons of spinal cord
- Interacts with HSPB1
- Z-disk-associated CASA complex
- Participates in chaperone-assisted autophagy
- Mutated protein promotes formation of intracellular aggregates
- Chaperone-related disorders
- Clinical
- Onset
- 14 to 35 years
- Weakness of toe extension
- Weakness
- Extensor muscles of feet (Large toe)
- Progression over 5 years to complete paralysis of all distal muscles of legs
- Legs > Arms
- Cramps & Fasciculations: Some patients
- Sensation: Usually normal
- Tendon reflexes: Reduced or absent in lower extremities
- Onset
- Laboratory
- Nerve conduction velocity: Normal
- EMG: Chronic denervation
- Serum CK: Mildly high
- HSPB8 variant syndrome: Motor neuropathy + Distal Myopathy
125
- Epidemiology: 2 families, 7 patients
- Genetics
- Inheritance: Dominant
- Mutations: c.421A>G (p.Lys141Glu); c.151insC
- Allelic disorders
- Clinical
- Onset age: 1st to 3rd decade
- Weakness
- Distal
- Legs > Arms
- Proximal with disease progression
- Cramps
- Fasciculations
- Tendon reflexes: Reduced in legs or Normal
- Sensation: Normal
- Scapular winging: Some patients
- Camptocormia: Some patients
- Course: Slow progression
- Laboratory
- NCV: Motor neuropathy
- Motor axon loss: Length dependant, Legs > Arms
- EMG: Distal leg denervation
- Muscle
- Fiber size: Varied
- Fiber type grouping
- Vacuoles: Rimmed & Non-rimmed; p62 & SMI-31 positive
- Aggregates: Desmin; Myotilin, αB-crystallin; Dystrophin
- Serum CK: 250 to 2,000
- Muscle MRI: Anterior leg involvement
- NCV: Motor neuropathy
- HSPB8 variant syndrome: MFM13 - Limb-Girdle Myopathy with Myofibrillar pathology & Rimmed vacuoles
161
- Epidemiology: 12 families
- Genetics
- Mutations
- Region: C-terminal
- Type: Stop; Frameshift
- c.577_580dupGTCA (p.Thr194Serfs*23); c.525_529del; c.576_579delinsCAG; c.562delC; c.520_523delTACT; c.515delC;
- Inheritance: de novo; Dominant
- Mutations
- Clinical
- Onset age: 6 to 37 years
- Weakness
- Legs > Arms
- Proximal: Common; Distal in some
- Symmetric
- Respiratory: Some patients; May be severe
- Trunk
- Paraspinous: Muscle wasting
- Scapular winging
- Cramps & Fasciculations: Some patients
- Tendon reflexes: Absent
- Sensation: Normal
- Contractures: Posterior neck; Rigid spine
- Cardiomyopathy: Some patients
- Course: Progressive
- Laboratory
- Serum CK: Normal to 1110
- EMG: Fibrillations; Small motor unit potentials
- Muscle imaging: Fatty replacement, Paraspinous & Thigh
- Muscle pathology
- Rimmed vacuoles
- Aggregates
- Myofibrillar proteins: Myotilin, Desmin, TIA1
- Autophagy related: p62; HSPB8, TDP-43
- Sarcolemma proteins: Dystrophin; Caveolin-3
- Lipid: Increase in type II fibers
- Cytoplasmic bodies
- Inflammation: Endomysial; Some patients
- Ultrastructure: Myofibrillar disorganization
Distal hereditary motor neuropathy (HMND3; HMN 2B)
●
Heat-shock 27-kD protein 1 (HSPB1; HSP 27)
- Epidemiology
- Common cause of dHMN
- Families from UK, Croatia, Belgium, Austria
- Genetics
- Missense mutations: R127W; S135F; T151I; P182L
- Mutations in C-terminal domain
- More severe phenotype
- Onset age: 4 to 7 years
- Allelic disorders
- Clinical
- Onset age: 21 to 54 years
- Weakness
- Legs: Distal; Early in course
- Arms: Distal; After 5 to 10 years
- Tendon reflexes: Reduced or Brisk
- Course: Slow progression
- MRI: Anterolateral lower legs relatively spared
Distal hereditary motor neuropathy with upper motor neuron signs 22
● Senataxin (SETX)- Epidemiology: Belgian, Austrian & English families
- Genetics
- Clinical
- Onset age
- 4 to 49 years
- Weakness
- Legs & Arms
- Distal: Intrinsic hands & ankles
- Bulbar: Normal
- Sensory: Usually normal
- Tendon reflexes: Normal or Increased
- Babinski sign: Positive in 50%
- Pes cavus (50%)
- Onset age
- Laboratory
- Electrophysiology
- Nerve conduction velocity: Normal or Mildly reduced
- CMAPs: Reduced amplitude
- Sensory: Normal SNAP amplitude; Conduction velocity borderline or mildly slow
- Central motor conduction latencies: Slow
- EMG: Distal denervation
- Sural nerve pathology: Minor changes
- MRI: White matter changes in 1 patient
- Electrophysiology
- Differential Diagnosis
Distal SMA: Upper limb predominance (HMND5; HMN 5A; SMAD1)
●
Glycyl tRNA Synthetase 1 (GARS1)
- Epidemiology: Multiple ethnic origins
- Genetics
- Allelic disorders
- Mutations: Missense; E71G, L129P, G240R, Leu272Arg, G526R
- Asymptomatic parent: May be mosaic\
- ARS mutations
- GARS protein
- Clinical
- Onset
- Age
- Mean 17 years
- Range: 7 months to 4th decade
- Weakness: Hands
- Age
- Weakness: Varied severity
- Thenar eminence & 1st Dorsal Interosseous
- Lower extremities involved in 50% after 2 years
- Severe cases: Axial; Respiratory
- Tendon reflexes: Reduced
- Cramps: Related to cold exposure or exercise
- Joints: Hyperlaxity
- Rare Pyramidal signs: Rare
- Progression: Very slow to
- Onset
- Laboratory
- NCV: Motor axon loss
- Muscle: SMA-like denervation
Distal SMA: Upper limb predominance (HMND13; HMN 5C)
6
●
BSCL2 gene (Seipin)
- Nosology: Also called HMN 5A
- Epidemiology
- European families
- Most common D-HMN
- Genetics
- Mutations
- Missense
- Exon 3
- Common: Asn88Ser; Ser90Leu
- Located at N-glycosylation site
- Mutation effects
- Alters N-glycosylation site
- Aggregate formation
- BSCL2 allelic disorders
- Mutations
- Seipin protein
- Integral membrane protein: Endoplasmic reticulum
- Glycosylated
- Lipid droplets: Synthesis & ER contacts
- Clinical
- Onset
- Age: Mean 15 to 24 years; Range 2 to 40 years; Childhood often
- Weakness: Hands
- Weakness
- Hands
- Early involvement
- Thenar eminence > 1st Dorsal Interosseous
- Asymmetric
- Lower extremities
- Peroneal weakness (60%)
- Symmetric
- Foot deformities (95%)
- Proximal: Normal
- Hands
- Upper motor neuron
- Tendon reflexes: Brisk
- Tone: May be increased in legs
- Plantar responses: Flexor
- Sensory loss: Vibration reduced in legs
- Hyperhidrosis (40%): Hands & Feet
- Progression
- Very slow: Over decades
- No patients severely handicapped
- Onset
- Laboratory
- Electrophysiology
- CMAPs: Small
- Motor NCV: Normal or Mildly reduced
- Sensory NCV amplitude: Mild reduction, especially older patients
- EMG: Large MUPs; Reduced Recruitment
- Central motor conduction times: Prolonged (60%)
- Sural nerve: Mild loss of myelinated axons
- Muscle MRI: Thenar eminence, Soleus & Tibialis anterior most involved
- Electrophysiology
- BSCL2 variant syndromes
- CMT2-like
99
- Genetics
- BSCL2 Mutation: S90W
- Inheritance: Dominant
- Clinical
- Onset ages: 5 to 30 years
- Weakness: Legs early; Thenar; Distal
- Sensory loss: Pan-modal; Distal
- Spastic: Gait; Tendon reflexes increased (Legs); Extensor plantar response
- Skeletal: Pes cavus
- NCV
- CMAPs: Small amplitudes, especially median
- SNAPs: Small amplitude in some
- Velocities: Normal
- Sensory myelinated axons: Loss or Increased; Regeneration; Thin myelin
- Genetics
- Also see
- CMT2-like
99
Hereditary Distal Ulnar-Median Muscular Atrophy 7
● Autosomal Dominant- ? HMN 5 variant
- Weakness
- Distal
- Arms at onset; Legs later
- Symmetric
- Onset: Childhood - Teens
- Upper motor neuron signs
- Brisk tendon reflexes
- Plantars reflexes: Flexor, Equivocal or Extensor
- Sensory: Normal
- Electrodiagnostic
- Normal nerve conduction velocities
- Prolonged distal latencies
Distal Hereditary Motor Neuronopathy 7A (HMND7; HMN 7A; dHMN-VII; dHMN7) (Vocal cord involvement)
●
Solute carrier family 5 (Choline transporter), Member 7 (SLC5A7; CHT)
- Epidemiology: 4 families
- Genetics
98
- Mutations
- Types: Frameshift or Tuuncating
- c.1497delG (p.Lys499Asnfs*13), p.His521Gln*fs2, p.Lys510Asnfs*2, c.1526del (p.Pro509Leufs*3)
- Allelic disorders
- Congenital MG with Episodic Apnea: Recessive, Missense mutations
- Lethal Congenital Arthrogryposis
- Mutations
- SLC5A7 protein
- Choline transporter
- Presynaptic: Motor neurons
- Determinant of synaptic acetylcholine synthesis & release at NMJs
- Mutation: Reduced choline transport; Remove endoytic trafficking motif
- Clinical
- Onset
- Age: Early childhood to Teens
- Voice or Gait disorder
- Distal weakness
- Onset: Hands; Median distributaion
- Progression to distal leg weakness
- Wasting: Prominent distally
- Usually symmetric but occcasional asymmetry
- Vocal cord involvement (70%)
- Distribution: Often asymmetric; Eventually bilateral
- Onset: 1st or 2nd decade
- Voice: Hoarse; Quiet
- Respiratory failure: 2° Bilateral vocal cord paralysis
- ± Sensorineural hearing loss
- Tendon reflexes
- May be brisk
- Reduced distally in arms & legs with disease progression
- Sensation: Normal
- Course: Slow progression
- Onset
- Electrodiagnostic
- EMG: Distal denervation in feet & hands
- NCV
- Velocities: Normal
- CMAPs: Small distally
- Repetitive nerve stimulation: No decrement
- SFEMG: Excess jitter
- SLC5A7 variant disorder: Congenital MG 20 (CMS20), Presynaptic, with Episodic Apnea
133
- Epidemiology: 6 families
- Genetics
- Inheritance: Recessive
- Mutations: Missense; Loss of function
- Allelic disorders
- Clinical
- Onset
- Age: Congenital to 2 months
- Apnea
- Weakness
- Bulbar: Dysphonia; Dysphagia
- Face
- Eye: Ptosis; Ophthalmoparesis
- Limbs: Proximal > Distal
- Axial: Neck & Other
- Variability
- Apnea: Episodic
- Fatigability
- Arthrogryposis
- Fingers & Knees
- In more severe syndromes
- Associated with: Hypotonia
- Intellectual disability
- Course: Fluctuating weakness; Improvement with treatment
- Treatment: AChE inhibitors
- Onset
- Laboratory
- Repetitive nerve stimulation decrement: 0% to 70%
- Neuromuscular junctions
- Young patient: Immature, Reduced definition of AChR patches; Polyinnervated
- Older patient: NMJs often Denervated or Remodeled; Butyrylcholinesterase (BChE) increased
Distal Hereditary Motor Neuronopathy (Vocal cord involvement) (HMND14; HMN 7B)
34
●
Dynactin 1 (DCTN1)
- Epidemiology: > 10 families
- Genetics
- Mutations: Missense most common; Gly59Ser (In CAP–Gly domain); R1150C
- DCTN1 allelic disorders
- Perry disease
- Mutations: CAP–Gly domain
- Parkinsonism, Depression, Weight loss, Hypoventilation, TDP-43 immunostaining of brain
- May be associated with
- ALS susceptibility: C-terminal, Non-CAP–Gly domain mutations
- Progressive supranuclear palsy (K56R)
- Early onset motor disease (c.626dupC)
- Perry disease
- Dynactin protein
- Largest subunit of 10 million dalton dynactin complex
- Binds to
- Microtubules
- Dynein: Cytoplasmic
- Also see: Rab proteins, CMT 2B
- Functions
- Associated with axonal transport of vesicles & organelles
- Blockade of dynactin binding to dynein: Blocks vesicle motility along microtubules
- May promote synapse stability at neuromuscular junctions
- Mutation
- Located in p150Glued subunit
- May distort folding of microtubule binding domain: Reduced tubulin binding
- Induces dynactin self-aggregation & with dynein (in vitro)
56
- Associated with mitochondria
- Aggregation reversed by overexpression of Hsp70
- Clinical
- Onset
- 3 years to 4th decade
- Respiratory difficulty due to vocal cord paralysis
- Weakness/Motor
- Face: Progressive; Fasciculations
- Vocal cords: Paralysis
- Tongue: Fasciculations
- Limbs: Distal > Proximal; Hands then Feet; "Split hand"
- Sensory: Normal
- Ankle contracture: With early onset
- Tendon reflexes: May be brisk or reduced
- Cognition: Normal
- Other
- Gynecomastia: Some patients
- Foot deformity
- Course: Slow progression
- Onset
- Laboratory
- NCV: Velocity normal, CMAPs small
- EMG: MUPs large & long
- Variant syndrome: ALS or ALS susceptibility
- Genetics
- Inheritance: Dominant or Sporadic
- Mutations: Missense; Thr1249Ile, Met571Thr, Arg785Trp, Arg1101Lys; R1275C
- Incomplete penetrance
- Clinical
- Onset
- Age: 46 to 64 years
- Weakness: Arms, Legs, Posterior neck or Bulbar
- Weakness
- Arms, Legs or Bulbar
- May be asymmetric
- Progression: Slow; symptom duration = 4 to > 9 years
- Upper motor neuron signs: Present
- Tendon reflexes: Brisk
- Fronto-Temporal Dementia
- Some patients without motor neuron disease
- 1 family
- Onset
- Laboratory
- EMG: Widespread denervation
- MRI: Normal
- Genetics
Distal SMA: Calf predominant (HMND6; HMN2D)
106
●
F-box only protein 38 (FBXO38; MOKA)
- Nosology: Neuronopathy, distal hereditary motor, type IID
- Epidemiology: 2 families
- Genetics
- Mutation: Cys206Arg
- Allelic disorder: dHMN, Recessive
- FBXO38 protein
- Coactivator of transcription factor Kruppel-like factor 7 (KLF7)
- Regulates genes required for neuronal axon outgrowth & repair
- Coactivator of transcription factor Kruppel-like factor 7 (KLF7)
- Clinical
- Onset
- Age: 13 to 48 years
- Difficulty standing or running
- Weakness
- Initial: Calves
- Distal predominant: Hands, Intrinsic; Feet
- Other weak muscles: Triceps
- Proximal: With disease progression in some patients
- Tendon reflexes: Ankle absent
- Fasciculations
- Course
- Slowly progressive
- Some patients lose ambulation
- Onset
- Laboratory
- NCV
- Motor amplitudes: Reduced
- Sensory: Normal
- EMG: Neurogenic; Fibrillations
- Muscle biopsy: Grouped atrophy; Large fibers types II > I
- NCV
- FBXO38 variant: Distal hereditary motor neuronopathy type (dHMN) IID, Recessive
156
- Epidemiology: Turkish patient
- Genetics
- Inheritance: Recessive
- Mutation: Homozygous; p.Arg526Gln
- Clinical
- Onset: Childhood
- Weakness: Distal; Legs > Arms
- Tendon reflexes: Normal
- Sensory: Normal
- Skeletal: Pes cavus
- Hearing loss
- Systemic: Duplex collective system, Arcuate uterus, Choanal atresia
- Laboratory
- EMG: Denervation, distal
- NCV: CMAP amplitudes small
- Brain MRI: Normal
Distal SMA: Leg predominant 19
● Dominant- Epidemiology: Single Italian family
- Onset: 8 to 30 years; Difficulty with heel walking
- Clinical
- Weakness
- Legs: Distal; Tibio-Peroneal
- Arms: Mild; Later in disease course
- Proximal: Mild; Arms & Legs; Late in course
- Hearing: Sensorineural loss in older patients
- Weakness
- Laboratory
- EMG: Denervation in distal muscles
- Nerve conduction: CMAPS small; Normal NCV
- Muscle biopsy: Chronic denervation
- Auditory evoked potentials: Cochlear hearing loss
- See: Congenital SMA of lower limbs
Distal SMA 3 (HMNR3; DSMA 3)
21
●
Chromosome 11q13.3; Recessive
- Nosology: HMN types III & IV
- Epidemiology
- Lebanese & European families
- Most patients probably from single ancestor
- Genetics: Normal IGHMBP2 gene
- Clinical
- Onset
- Age: Infancy to Early adult
- Distal weakness
- Weakness & Atrophy
- Distal
- Feet > Hands
- Diaphragm: With childhood onset
- Proximal & Trunk: With disease progression
- Cranial nerves: Normal
- Sensation: Normal
- Tendon reflexes: Reduced or Normal
- No upper motor neuron involvement
- Course: Slow progression
- Onset
- Electrophysiology
- EMG: Denervation
- NCV: Normal
- Muscle biopsy: Denervation
Distal HMN: Childhood onset
●
Autosomal Recessive
- Clinical features
- Onset: Early childhood
- Weakness: Distal; ± Quadriceps
- Progression: Very slow; Survival until at least middle life
Distal infantile spinal muscular atrophy with diaphragm paralysis (HMNR1; DSMA1; SMARD1; HMN 6)
●
Immunoglobulin μ-binding protein 2 (IGHMBP2)
- Epidemiology
- > 50 patients
- Up to 1% of early onset SMA
- IGHMBP2 genetics
- Mutations: Missense; Nonsense; Frameshift deletion (exon 5) & Splice donor-site
- Allelic disorders
- DSMA1 (SMARD1; HMN6)
- SMARD, milder
- AR-CMT2S
- IGHMBP2 protein
66
- Transcription factor: DNA binding protein
- ATP-dependent 5' --> 3' helicase: Separates double-stranded RNA & DNA
- Regulates: DNA replication, Pre-mRNA splicing, Transcription ± Translation
- Localization
- Similar to SMN1 protein
- Co-localization
- RNA-processing machinery in both cytoplasm and nucleus
- Ribosomes
- Tissue distribution: Widespread
- Mutations: Impair ATPase & Helicase activity
- Clinical features
- Onset
- Age: Congenital to 2 years
- Intrauterine growth retardation
- Respiratory failure
- Hypotonia
- Respiratory distress: Severe
- Diaphragmatic paralysis
- Intercostal muscles: Relatively spared
- Weakness: Predominantly upper limbs & distal muscles
- Tendon reflexes: Reduced
- Contractures: Occasional; Mild; At knee and ankle
- Fingers: Fat
- Course
- Common: Death or respiratory failure at < 3 months
- Onset
- Serum CK: Normal
- Chest x-ray: Eventration of diaphragm
- Spinal cord pathology
- Upper more severely affected than the lower
- Small anterior roots
- Remaining motor neurons show chromatolysis
- Nerve (Sural): Myelinated axon loss
- Muscle
- Neurogenic atrophy without reinnervation
- Large fibers: Type I
- Genetics
- May have same mutation & family as severe disease
- Clinical
- Onset: 2nd year
- Weakness
- Distal then Proximal
- Dysphagia
- Respiratory
- Sleep hypoventilation
- Vital capacity: Normal
- Survival through childhood
- Epidemiology
- 12% of AR-CMT2
- Asian, European, US
- IGHMBP2 Genetics
- Inheritance: Recessive
- Mutations: Heterozygous; Missense, Stop or Splice; Common p.Cys46*
- Clinical
- Laboratory
- NCV: Axon loss, Motor & Sensory
- Nerve biopsy: Loss of large axons
- Splice donor mutation
- Functional IGHMBP2 expression reduced to 20–25% of controls
- Life spans: 12 to 138 days
- Weakness & Muscle wasting
- Progressive & Severe
- Hind limbs then fore limbs
- No phrenic nerve or diaphragm involvement
- Loss of motor neuron innervation
- Genetic disease modifier: On mouse chromosome 13
- Other tissues involved if IGHMBP2 expression replaced in nerve
- Dilated cardiomyopathy: High serum CK & CK-MB
- Skeletal muscle: Myopathy, mild
Distal Hereditary Motor Neuropathy, Jerash type (DSMA 2; HMNJ)
227
●
Sigma-1 Receptor (SIGMAR1; σ1R)
- Epidemiology: 10 families
- Genetics
- Locus contains: SIGMAR1 gene
- Clinical
- Onset age: 2 to 10 years
- Weakness
- Distal
- Legs, then Arms within 2 years
- Muscle wasting: Hands & Feet
- Steppage gait
- Tendon reflexes
- Brisk at Knees; 6 to 25 years
- Ankle: Absent
- Upgoing toes: Younger than 15 years
- Laboratory
- Electrophysiology
- Motor: Small CMAPs; Normal NCV
- Sensory: Normal SNAPs
- EMG: Chronic denervation
- Serum CK: Normal
- Biopsy: Sural Nerve
- Mild reduction in number of myelinated axons
- Occasional regeneration
- Biopsy: Muscle
- Grouped atrophy
- Target fibers
- Type I predominance
- Electrophysiology
Distal SMA, X-linked 3 (SMAX3)
41
●
ATPase, Cu++-transporting, alpha polypeptide (ATP7A)
- Epidemiology: Brazilian, Australian & North American families
- Genetics
72
- Mutations
- Missense
- Locations: P1386S; T994I
- Allelic disorders
- Menkes disease
: Loss of function mutations
- Occipital horn syndrome
- Defects that allow residual copper transport
- Often via leaky splice-junction mutations involving noncanonical bases
- Menkes disease
- Mutations
- ATP7A protein
- Copper-transporting P-type ATPase
- Normal copper: Localizes to trans Golgi network in basal copper concentrations
- Elevated copper: Relocates to small vesicles and plasma membrane
- Mutations
- ATP7A protein levels: Normal
- Functions: Impaired ATP7A trafficking in response to copper loading
- Clinical
- Onset
- Age: 1 to 61 years; Older in Australian family
- Foot deformity
- Gait disorder
- Weakness & Atrophy
- Legs
- Early in disease course
- Distal
- Tibioperoneal
- Hands
- Usual: With disease progression
- Early in disease course: 1 patient
- Proximal: Normal
- Temperature sensitive: Occasional patients weaker in cold
- Progression
- Slow
- Ambulation: Remains independent
- Legs
- Tendon reflexes: Variable; Absent diffusely or in legs; May be normal
- Plantar response: Neutral
- Skeletal: Pes cavus
- Autonomic: Normal
- Hair: Normal
- Joints & Skin: Normal
- Heterozygous females: Normal
- Onset
- Laboratory
- Serum CK: Normal
- Electrophysiology
- CMAP amplitudes: Reduced or Absent
- NCV: Mildly reduced or normal
- EMG: Chronic denervation
- SNAPs: Normal
- Pathology
- Nerve (Sural): Normal
- Muscle: Denervation
- Normal
- Catecholamine ratios in plasma
- β-2-microglobulin in urine
Spinal muscular atrophy with respiratory failure (SMARD) 2, X-linked (SMAX) 111
● LAS1-like Ribosomal biogenesis factor (LAS1L)- Epidemiology: 2 patients
- Genetics
- Mutations: S477N, Splice (c. 846 G>C)
- Allelic disorder: Wilson-Turner syndrome
- LAS1L protein
- Function: Ribosomal biogenesis
- Coordinates processing of 45S rRNA at both ends of 2nd internal transcribed spacer
- Clinical
- Onset age: Neonatal
- Weakness
- Distal
- Respiratory failure: Early onset; Diaphragm paralysis
- Feeding difficulty
- Fasciculations: Tongue
- Hypotonia
- Contractures: Mild; Toes & Fingers
- Tendon reflexes: Present
- SMARD: Differential diagnosis
- Laboratory
- Electrodiagnostic
- CMAP amplitudes: Reduced in legs more than arms
- SNAPs: Normal
- EMG: Reduced recruitment, especially distal
- Brain MRI: Normal
- EEG: Normal
- CSF: Normal
- Electrodiagnostic
Distal hereditary motor neuropathy 45
● Chromosome 11p; Recessive- Epidemiology: Southern Italian family
Distal Hereditary Motor Neuropathy (HMND4; HMN 2C)
71
●
Heat-shock 27-kd protein 3 (HSPB3; HSPL27)
- Epidemiology: 4 families
- Genetics
- Mutations: Missense; Arg7Ser; R116P; Y118H
- Allelic disorder: Shoulder girdle weakness, p.L34Ffs*50 (A33AfsX50)
- Other: Small heat-shock protein disorders
- HSPB8: HMN2A & CMT 2L
- HSPB1: HMN2B & CMT 2F
- αB-crystallin: Myofibrillar myopathy & Posterior polar cataracts
- HSF1 antibodies
- HSPB3 protein
- Clinical
- Onset
- Age: 3rd decade
- Weakness: Legs
- Weakness
- Distal
- Legs: Severe at ankles
- Arms: Hands with wasting
- Shoulder-girdle: Stop mutation, heterozygous
- Sensation: Normal or Mildly reduced
- Tendon reflexes: Reduced at ankles
- Onset
- Laboratory
- EMG: Denervation in distal legs & hands
- NCV: Normal velocities; Axon loss
Distal Hereditary Motor Neuropathy with Pyramidal features
● Chromosome 4q34.3-q35.2; Dominant- Epidemiology: Italian family
- Clinical
- Onset
- Age: 25 to 40 years
- Gait Disorder: Spastic
- Weakness & Atrophy
- Distal
- Legs
- Upper motor neuron
- Legs
- Spasticity
- Tendon reflexes: Brisk at knees; Reduced at ankles
- Sensation: Slight vibratory loss in feet
- Cognition & Bulbar: Normal
- Progression: Slow; Walking preserved
- Onset
- Laboratory
- Motor nerve conductions: Velocity normal; Amplitude reduced
- SNAPs: Normal
Spinal Muscular Atrophy: Other
Infantile Spinal Muscular Atrophy with Arthrogryposis (XL-SMA; SMAX2; AMCX1)
● Ubiquitin-activating enzyme 1 (UBE1; UBA1)
- Epidemiology: 8 families
- Genetics
- UBE1 (UBA1) protein
168
- Expression: All tissues; High levels in motor neurons
- Localization
- Nuclear in normal motor neurons
- Cytoplasmic SMN1-linked SMA motor neurons
- Ubiquitin-proteasome system
- Ubiquitin activating E1 enzyme
- Initiates activation & conjugation of ubiquitin-like
proteins
- Binds to gigaxonin
- Modification of proteins with ubiquitin or ubiquitin-like proteins
- Controls many signaling networks
- Requires
- Ubiquitin activating enzyme (E1)
- Ubiquitin conjugating enzyme (E2)
- Ubiquitin protein ligase (E3)
- Ubiquitin activating enzyme (E1)
- Required for Atg7- & Atg3-independent autophagy
- Clinical features
- Onset: Congenital or Infant
- Weakness
- Early: Hypotonia
- Proximal > Distal: Similar to Werdnig-Hoffmann
- Face: Myopathic faces
- Bulbar
- Speech: Nasal
- Tongue: Fasciculations
- Cramps: Some patients
- Tendon reflexes: Reduced
- Skeletal
- Contractures: Proximal & Fingers; Congenital
- Fractures: Congenital
- Face: Dysmorphism
- Genital: Undescended testes
- Course
- Severe disorders: Usually death < 2 years
- Associated with respiratory insufficiency
- Milder patients: Long lifespan
- Family history: Miscarriages/spontaneous abortions
- Laboratory
- Pathology
- Muscle: Denervation
- Anterior horn cell loss
- Brain MRI: Normal
- Serum CK: Mildly high
- NCV: CMAP amplitudes reduced; Velocities normal
- EMG: Fibrillations; Positive sharp waves
- Pathology
- Female carriers: No symptoms
- Rule out: Congenital 5q-linked SMA
- UBA1 Allelic disorder: VEXAS
216
- Nosology
- VEXAS: Vacuoles, E1 enzyme, X-linked, Autoinflammatory, Somatic
- Epidemiology: > 50 years of age
- Male: 1 in 4,269
- Female: 1 in 26,238
- Genetics
- Mutations: Somatic; Met41Val, Met41Thr, Met41Leu
- Mutation locations: Hematologic stem & progenitor cells
- Clinical
- Males > > Females
- Onset age: 7th & 8th decades
- General
- Autoinflammatory, Treatment-resistant syndromes
- Fever
- Weight loss
- Skeletal: Chondritis, Joints
- Skin: Neutrophilic dermatosis, Leukocytoclastic vasculitis
- Thrombosis
- Pulmonary infiltrates
- Ocular: Diplopia; Scleritis; Uveitis
- Vasculitis (24%)
- Skin (2%)
- Small (19%) > Large vessels (2%)
- Neutrophil inflammation
- Some ANCA+
- Not: Giant cell arteritis
- Other: Pericarditis; Renal; Testicular; GI
- Neurologic (6% to 14%)
- 1.5 to 4 years after disease onset
- Acute onset (< 4 weeks)
- Polyneuropathy
- Clinical
- Sensory ± Motor
- Symmetric or Asymmetric
- Limbs or Cranial nerves
- Some: Multiple mononeuropathies
- Laboratory
- NCV: Axon loss
- Pathology: Axon loss; Some with demyelination/CIDP
- CSF protein: Commonly high
- Clinical
- Cranial nerves: III; VI
- CNS
- Encephalopathy
- Infarction
- PRES
- Aseptic meningitis
- Treatments
- Corticosteroids
- Other: Ruxolitinib, Azacitidine, Tocilizumab
- Hematopoietic stem cell transplantation
- Course: 30% mortality at 4 years
- Laboratory
- Hematologic: Cytopenia, Macrocytosis, Vacuoles in myeloid & erythroid precursors, Bone marrow dysplasia
- Nosology
- Autoinflammatory syndromes, Other
Proximal SMA with Dominant inheritance: Adult Onset (Finkel, Late adult type; SMAFK)
● VAPB
- Epidemiology
- Common in Brazil; > 200 patients
- Other families: German, Dutch, France, China
- Genetics
- Brazil & Portugal mutation: P56S
- Allelic disorder: ALS 8
- Clinical
- Onset
- Age: 30 to 60 years; Mean = 49 years
- Legs
- Weakness & Atrophy
- Proximal
- Respiratory: Some patients
- Gait: Waddling
- Fasciculations
- Tendon reflexes: Reduced (80%)
- Sensory & Bulbar: Normal
- Course: slow progression
- Onset
- Laboratory
- Serum CK high
- EMG: Neurogenic; Positive sharp waves, MUPs long duration & large amplitude
- NCV: Velocities normal; CMAP amplitudes reduced; SNAPs normal
- Muscle biopsy: Neurogenic
Hereditary motor neuropathy, distal 8 (HMND8; HMN8): Spinal muscular atrophy, Congenital, non-progressive, of lower limbs (CSMAA)
● TRPV4
- Epidemiology: Multiple families
- Genetics
- Mutations: Missense; Ser94Leu, Arg269Cys (Also with CMT 2C & SPSMA)
- Allelic disorders
- Clinical
- Onset age: Congenital
- Weakness
- Developmental delay: Motor
- Legs only: Proximal & Distal
- Non-progressive
- Vocal cord paralysis: Some patients
- Tendon reflexes: Reduced in legs
- Contractures
- Arthrogryposis
- Knees & Ankles
- Laboratory
- Serum CK: Mildly elevated
- Electrophysiology
- EMG: Chronic (Giant motor units) & Active denervation
- NCV: Normal motor & sensory
- Differential diagnosis
- Distal SMA of lower limbs
- SMALED: DYNC1H1
- SMALED2: BICD2
- SMA Congenital, non-progressive, of lower limbs: TRPV4
Hereditary Motor Neuropathy, Distal 9 (HMND9; HMN9; dHMN type I)
● Tryptophanyl-tRNA synthetase 1 (WARS1)
- Epidemiology: 5 families; 14 patients
- Genetics
- Mutation: c.770A>G (p.His257Arg) (Recurrent); Phe138Tyr (de novo); Asp314Gly (Older onset)
- Allelic disorder: Microcephaly + Developmental delay & Brain anomalies, Recessive 187
- WARS1 protein
- Function
- Cytoplasmic Amino-acyl tRNA synthetase (ARS)
- Catalyzes aminoacylation of tRNAtrp with tryptophan
- Mutation effect: Dominant negative
- Function
- Clinical
- Onset
- Age: 9 to 23 years
- Weakness: Distal; Legs
- Gait disorder
- Weakness & Wasting
- Distal
- Arms & Legs
- Moderate to Severe
- Sensation: Normal
- Tendon reflexes: Ankles absent; Knees absent or normal
- Feet: High arches
- Course
- Progressive
- Most remain ambulant
- 1 patient in wheelchair
- Onset
- Laboratory
- Electrodiagnostic
- NCV: 35 to 63 M/s
- CMAPs: Reduced amplitude
- SNAPs: Normal amplitude
- Pathology
- Sural nerve: Normal
- Muscle: "Neurogenic atrophy"
- Muscle MRI: Distal atrophy
- Electrodiagnostic
Early-onset spinal muscular atrophy with Contractures (SMALED2A)
● Bicaudal D, drosophila, homolog of, 2 (BICD2)
- Epidemiology: 23 families
- Genetics
- Mutations
- Hot spot: Ser107Leu
- Other: Asn188Thr; Ile189Phe; Val485Gly; Arg501Pro; Lys508Thr;
Ala535Val; Tyr557His; Ser681Leu; Thr703Met; Arg747Cys; Glu774Gly - Locations: Coiled-coil domains
- Allelic disorders
- SMALED2A
- SPG
- Arthrogryposis (SMALED2B)
- Distal myopathy
- Mutations
- BICD2 protein
- Golgin
- Adaptor protein
- Maintains Golgi structure
- Interacts with the dynein-dynactin motor complex: Axon transport
- Facilitates trafficking of cellular cargos involved in motor neuron development & maintenance
- Mutations
- Increased microtubule stability
- Abnormal collateral axon branching
- Impaired NMJ development
- Dynein hyperactivity
- Dynein-related disorders, Other: SMA-LED; CMT 2O
- Clinical
- Onset
- Age: In utero, Congenital to Adult
- Fetal movements reduced
- Walking: Late, Difficult or Slow
- Contractures: Ankles
- Weakness
- Legs > Arms
- Legs: Proximal & Distal
- Arms: Mild; Proximal & Hands
- Respiratory: Sleep disordered breathing in some patients
- Symmetric
- Most patients ambulant
- Muscle wasting: Legs
- Skeletal
- Feet: Pes planus or High arch; Calcaneovalgus
- Hip dislocation: Congenital or Early-onset
- Spine
- Scoliosis (50%): Not severe
- Lordosis
- Contractures
- Common: Not all patients
- Locations: Knees; Ankles (Pes equinovarus)
- Onset: Congenital or 1st decade
- Scapular winging: 40%
- Upper motor neuron
- Spastic paraplegia: Some families; Later onset
- Tendon reflexes
- More severe disease: Brisk in legs or arms
- Reduced or absent in legs: Other patients
- Especially at ankles
- Fasciculations: Arms in some patients
- Sensation: Usually normal; Occasional vibratory loss
- Cognitive: Normal
- Course
- Slow progression or Stable
- Many remain ambulatory to 5th decade
- May need wheelchair
- Onset
- Laboratory
- MRI: Muscle
- Pelvis: Gluteus medius & minimus
- Thigh
- Involved: Anterior; Vastus lateralis, intermedius; Sartorius > Rectus femoris
- Spared: Medial adductors & Semitendinosus
- Leg: Posterior calf (Gastrocnemius)
- Similar to DYNC1H1-associated DCSMA
- Electrodiagnostic
- EMG
- Denervation with Reinnervation, Chronic
- No fibrillations
- NCV: Normal
- EMG
- Serum CK: Usually normal; Up to 1100
- Spinal cord pathology
- Motor neurons reduced: Lumbar more than cervical
- Ventral roots: Atrophy
- Muscle pathology
- Chronic denervation
- Type 1 predominance & hypertrophy
- Groups of fast myosin negative muscle fibers
- Fiber size: Varied
- Pseudomyopathic
- MRI: Muscle
- SMALED: Differential diagnosis
- BICD2 variant syndrome: Spastic paraparesis (SPG)
- Epidemiology: 1 family, 4 patients
- Genetics
- Inheritance: Recessive
- Mutations: Especially coiled-coil domain 2; Missense; c.G1823A, p.S608L
- Allelic with: SMALED2
- BICD2 protein
- Clinical
- Onset age: 15 to 18 months
- Spasticity: Gait disorder; Tendon reflexes brisk; Plantar reflex extensor
- Amyotrophy (50%)
- Sensation: Normal
- Cognition: Normal
- Laboratory
- Brain MRI: Normal
- EMG/NCV: Normal
- BICD2 variant syndrome: Arthrogryposis
(SMALED2B)
135
- Epidemiology: 7 sporadic patients
- Genetics
- Inheritance: de novo; Dominant
- Mutations: Gln194Arg, Cys542Trp; Arg694Cys
- Clinical
- Fetal: Hypokinesia
- Skeletal
- Fractures, congenital
- Hip dislocation
- Micrognathia
- Arthrogryposis
- Hypotonia
- Respiratory insufficiency
- Microcephaly
- Cognitive delay
- Muscle: Atrophy
- Course: Early death in some
- Laboratory
- Brain imaging: Perisylvian polymicrogyria; Cerebellar vermis hypoplasia; Corpus callosum thin
- Muscle: Neurogenic atrophy
- EMG: Neurogenic
- BICD2 variant syndrome: "Distal myopathy"
- Epidemiology: 3 families
- Genetics
- Inheritance: Dominant
- Mutations: Arg622Trp
- Clinical
- Onset age
- Weakness
- Symmetric
- Distal legs: Especially foot dorsiflexors
- Laboratory
- Muscle MRI pathology: Thigh anterior & medial; Leg, Anterior > Posterior
- Serum CK: Mildly high
- EMG: Mixed myopathic & Neuropathic
- NCV: Normal Motor & Sensory
- Muscle pathology: Fiber size varied; Endomysial connective tissue increased
Spinal muscular atrophy with Lower limb predominance (SMA-LED)
● Dynein, cytoplasmic 1, heavy chain 1 (DYNC1H1)
- Epidemiology: > 10 patients
- Genetics
- Mutations: Missense; Arg251Cys, I584L, K671E, Y970C; Tail domain & other
- Allelic disorders
- DYNC1H1 protein
- Other dynein related disorder: Congenital SMA with contractures (SMALED2)
- Clinical
- Onset
- Age: Early childhood
- Leg weakness
- Late walking
- Weakness
- Most severe: Quadriceps & Psoas; Hip abduction
- Other: Distal legs; Mild
- Arms: Normal
- Symmetric
- Functional disorders
- Difficulty climbing stairs & rising from chair
- Waddling gait
- Course: Non-progressive or Progressive in episodes
- Wasting: Quadriceps & Distal legs; Small hand muscles
- Tendon reflexes: Reduced at knees; Others normal
- Sensation: Normal
- Contractures: None
- CNS: Learning difficulty in some patients; Seizures
- Onset
- Laboratory
- Muscle MRI: Thigh
- Early involvement: Vastus lateralis; Sartorius
- Hypertrophy: Adductor longus; Semitendinosus
- Electrophysiology
- EMG: Chronic denervation
- Potentials: Large amplitude, long-duration
- No spontaneous activity
- Distribution: Proximal & Distal Legs; Not paraspinous
- NCV: Normal velocities
- EMG: Chronic denervation
- Muscle biopsy
- Denervation: Small angular muscle fibers
- Type 2 muscle fiber predominance
- Focal inflammation: Perivascular
- Chronic pathology: Muscle fibers small; Endomysium increased
- Muscle MRI: Thigh
- Differential diagnosis
Hereditary Motor Sensory Neuropathy, Proximal (HMSNO; HMSN-P; CMT 2G)
● Trk-fused Gene (TFG)
- Epidemiology: Okinawa, Japan, Brazil, Taiwan (CMT2), Korea families
- Genetics
- TFG protein
- Endoplasmic reticulum exit sites
- Vesicles: Trafficking & Biogenesis
- COPII-mediated export
- Secretory cargoes
- ER → ER-Golgi intermediate compartments
- Inhibition: Slows protein secretion from endoplasmic reticulum (ER); Altered ER morphology
- Full length transcript: Predominantly expressed in neural tissues
- Clinical
- Onset
- Age: Weakness commonly in 5th decade; Range 17 to 50 years
- Cramps: Painful; Onset in 3rd decade
- Fasciculations
- Motor
- Weakness
- Proximal > Distal
- Lower > Upper
- Symmetric
- Cramps
- Fasciculations
- Muscle atrophy
- Course: Progression to severe disability & wheelchair
- Weakness
- Tendon reflexes: Absent
- Sensory
- Loss: Vibration & Joint position > Pain
- Dysesthesias: Distal
- Tremor
- Diabetes Mellitus: 40% with hyperglycemia
- Onset
- Lab
- Hyperlipidemia: 25%
- Hyperglycemia: 30%
- Serum CK: High; 200 to 500
- CNS MRI: Normal
- Electrodiagnostic testing
- CMAP amplitude: Reduced
- SNAP amplitude: Reduced or Absent
- NCV: Velocity normal
- EMG: Denervation: Fasciculations
- Pathology: Spinal cord
- Decreased numbers of anterior horn cells
- Marked loss of myelinated axons in posterior columns
- Neuronal cytoplasmic inclusions: TDP-43, TFG, Optineurin & ubiquitin
- Golgi fragmentation
- Sural nerve
- Myelinated axons: Reduced numbers
- Ultrastructure: Aggregated ER; Mitochondria small
- Muscle
- Fiber size variability
- Internal nucle
- Fiber splitting
- Core-like or targetoid structures
- Grouped atrophy
- Fiber type groups
- Aggregates: TFG, TDP-43, p62
- TFG variant disorder: SPG 57
- Epidemiology: Sudan, Indian & Italian families
- Genetics
- Inheritance: Recessive
- TFG mutations: Arg22Trp, Ile66Thr, Arg106Cys, R106H
- Allelic disorders
- TFG protein
- Mutation
- Inability to self-assemble into oligomeric complex
- Abnormal secretion from ER
- Impaired axon fasciculation after neuronal differentiation
- Mutation
- Clinical
- Onset age: < 2 years
- Spasticity
- Legs > Arms
- Gait: No independent walking
- Tendon reflexes: Increased
- Eye
- Optic atrophy: Reduced visual acuity
- Polyneuropathy
- Distal wasting: Hands & Feet
- Sensation: Normal
- Cognitive: Normal
- Laboratory
- NCV: Polyneuropathy
- Axon loss
- Demyelinating features: NCV reduced; Latencies increased
- Sensory: SNAP amplitudes reduced
- Motor: CMAPs reduced amplitude or Absent
- Brain MRI: Normal
- Nerve pathology: INAD
- CNS: Length dependent corticospinal tract axonopathy
- NCV: Polyneuropathy
- Diferential diagnosis: SPOAN
- TFG variant disorder: SPG, Dominant
200
- Epidemiology: 1 family, 5 patients
- Genetics
- Inheritance: Dominant
- Mutation: Missense; R42Q; Heterozygous
- Mutation effect: Autophagy defect
- Clinical
- Onset age: Early Childhood
- Spasticity
- Legs
- Gait disorder
- Progressive
- Weakness: Legs, Proximal ± Distal
- Laboratory
- Brain MRI: Normal
- NCV: Normal
- EMG: Denervation in legs
- Muscle pathology: Fiber type groups; Grouped atrophy
- TFG variant disorder: CMT2
160
- Epidemiology: Taiwan, Iranian & Italian families
- Genetics
- Mutation: Gly269Val
- Inheritance: Dominant
- Clinical
- Onset age: 28 to 40 years
- Weakness
- Distal
- Arms ≥ Legs
- Cramps
- Sensory
- Loss: Distal; Arms & Legs; Panmodal
- Paresthesias
- Gait ataxia
- Pain: Allodynia; Face
- Tendon reflexes: Reduced in legs
- Course: Progressive; May need walking aids
- Laboratory
- NCV: 44 to 61 M/s; Motor sensory axon loss
- EMG: Chronic denervation
- Nerve pathology: Axon loss, Large myelinated
Spinal Muscular Atrophy, Prenatal onset + Congenital Bone Fractures (SMABF)
● SMABF1
● SMABF2
- Epidemiology
- SMABF1: 16patients
- SMABF2: 13 patients
- Genetics
- Mutations: Stop; Often homozygous
- TRIP4 allelic variant: Congenital muscle disease
- ASCC1 variant: Congenital weakness, Developmental delay, Ulnar epiphysiolysis
- TRIP4 & ASCC1 proteins
- Expressed in embryos: Spinal cord, Brain, Paraspinal ganglia (Dorsal & Sympathetic), Thyroid & Submandibular glands
- ASC-1 transcriptional cointegrator complex
- Subunits: TRIP4; ASCC1; ASCC2; ASCC3
- Ribonucleoprotein complex
- Participates in: Transcriptional coactivation, RNA processing events
- ASC-1 binding protein: Cysteine and glycine rich protein 1 (CSRP1)
- Role in late myogenesis
- Expressed at low level in muscles, especially axial
- TRIP4 & ASCC1 location: Nucleus
- Clinical
- Genetic correlations
- ASCC1 mutations: More fractures; Shorter survival
- Pregnancy: Reduced fetal movements
- Motor
- Hypotonia
- Respiratory difficulty
- Diaphragm eventration
- Pulmonary hypoplasia
- Weakness
- Muscle mass: Decreased
- Tendon reflexes: Absent
- CNS: Global developmental delay
- Bone
- Generalized osteopenia
- Congenital fractures: Multiple; Long bones
- Joints: Arthrogryposis
- Multiple contractures: Proximal & Distal
- Congenital heart defects
- Secundum atrial septal defect
- Patent ductus arteriosus
- Cardiomyopathy
- Other: Some patients
- Face dysmorphism
- Microretrognathia,
- Hypertelorism
- High-arched palate
- Hypertrichosis
- Face dysmorphism
- Course: Death at 2 to 16 months
- Genetic correlations
- Laboratory
- EMG
- Neurogenic
- Muscle: No contraction to stimulation
- Pathology
- Muscle
- Fiber size: Atrophy; Variable
- Type I fibers: Larger; Clustered
- Fiber immaturity: Immature myosin (Developmental)
- Western blot: TRIP4 with reduced size, Upregulation of splice isoform
- Sural nerve: Loss of unmyelinated axons; Collagen pockets
- Spinal cord
- Anterior horn cells: Loss; Apoptosis
- Astrogliosis
- Muscle
- Brain MRI: Cortical gyration abnormal
- EMG
- TRIP4 variant: Congenital muscle disease (MDCDC)
132
- Nosology: Congenital muscular dystrophy, Davignon-Chauveau type
- Epidemiology: 6 families
- Genetics
- Inheritance: Recessive
- Mutations: c.G950A, p.W297*; Stop, Splice, Missense (Milder phenotype)
- Clinical
- Onset ages: Birth to adult
- Motor
- Hypotonia: Neonatal; Axial
- Respiratory failure
- Feeding difficulties
- Motor development: Delayed
- Weakness: Severe; Especially Proximal & Trunk
- Skeletal: Some
- Joint hyperlaxity
- Scoliosis: May be severe
- Rigid spine
- Contractures: Mild or None
- Dysmorphic: Flat face; Thick neck
- Skin
- Hyperelasticity
- Dry with scratch lesions
- Follicular hyperkeratosis
- Xerosis
- Cardiac: Dilated; Some patients
- Ophthalmoplegia: 2 patients
- Course
- Death in 1st years: Some
- Late: Non-walking
- Often stable
- Laboratory
- Serum CK: Normal or Mildly high
- EMG: Myopathic
- Muscle CT: Preservation of leg adductors
- Muscle MRI: Posterior thigh involvement with semi-tendinosus sparing
- Muscle biopsy
Pontocerebellar hypoplasia with Spinal muscular atrophy (PCH1A)
● Vaccinia-related kinase 1 (VRK1)
- Epidemiology: 3 families
- Genetics
- Mutations: Nonsense or Missense 68
- Allelic disorders
- Motor Neuron Disease, Distal ± Sensory loss
- ALS
- HMSN
- Microcephaly
- Spastic paraplegia
- VRK1 protein
- Serine/Threonine kinases
- Phosphorylates p53, c-Jun, CREB, ATF2, H2AX, NBS1, 53BP1
- Localization: Nuclear + Some cytoplasm & cell membrane
- Role in: Nuclear envelope formation
- Expression: Ubiquitous including brain
- Clinical
- Onset
- Age: 1st decade
- Severe cases: Prenatal or Birth; Death within months
- Milder cases: < 6 months
- Fetal movements: Reduced
- Hypotonia
- Feeding difficulty
- Age: 1st decade
- Motor
- Hypotonia
- Weakness
- Early: Reduced movements
- Diffuse
- Feeding disorders
- Progressive: Walking achieved in some then lost
- Milestones: Delay & Decline
- Polyneuropathy: Distal, Symmetric
- Tendon reflexes: Reduced or Increased
- CNS
- Cerebellar: Ataxia; Nystagmus
- Psychomotor retardation: Some patients
- Skeletal
- Contractures: Variable;
- Arthrogryposis: Severe cases
- Mild: Later onset patients
- Scoliosis
- Contractures: Variable;
- Course: Survial ranges from months to 12 years
- Onset
- Laboratory
- Serum CK: Normal
- MRI
- Cerebellar hypoplasia or absence, including vermis
- Pontine hypoplasia: Severe cases
- Microcephaly
- EMG: Neurogenic
- NCV: Axonal sensory-motor neuropathy
- Motor & Sensory velocities: Normal or Mildly slowed
- CMAP & SNAP amplitudes: Small
- Pathology
- Muscle: Denervation; Fiber type grouping; Grouped atrophy
- Nerve: Loss of large myelinated & small unmyelinated axons in sural nerve
- CNS
- Spinal cord: Motor neuron loss
- Cerebellum: Hypotrophic; Absent dentate nucleus; Other structural defects
- Brainstem: Hypotrophic; Posterior fossa cysts
- Basal ganglia: Neuronal loss
- Cortical atrophy
- VRK1 variant syndrome: Motor Neuron Disease, Distal ± (HMNR10)
124
- Epidemiology: 16 patients
- Genetics
- Inheritance: Recessive
- Mutations
- Missense > Stop
- Compound heterozygous
- H119R; G135R; Arg151Gln; L195V; V236M; R321C; R358X
- Clinical
- Onset age: 3 to 56 years; Mean 27 years
- Weakness Distribution
- Distal
- Legs (especially posterior) > Arms: Ankles & Toes; Hands
- Symmetric
- Respiratory
- Gait disorder
- Course: Slow progression over years
- Weakness Distribution
- Muscle
- Cramps
- Atrophy: Distal legs, posterior
- Tendon reflexes: Brisk, except absent at ankles
- Sensory loss (30%): Small fiber modalities in legs
- Skeletal: Pes cavus; Hammer toes
- Onset age: 3 to 56 years; Mean 27 years
- Laboratory
- Serum CK: High (347 to 3,400) or Normal
- NCV
- Motor: CMAPs small
- Sensory: Normal
- EMG
- Distal legs: Active denervation (Fibrillations)
- Arms & Proximal legs: Chronic denervation with reinnervation (Large motor units)
- Muscle biopsy: Small angular muscle fibers; Nuclear clumps
- Muscle MRI
- Fat infiltration
- Muscle Islands preserved
- Posterior compartment predominance
- Muscles: Gastrocnemius, Biceps femoris short head
- Brain MRI: Normal
- VRK1 variant: ALS
232
- Epidemiology: 20 patients
- Genetics
- Inheritance: Recessive
- Mutation: Asn234Asp, Arg321Cys
- Clinical
- Onset age: 3 to 38 years; Mean 15 years
- Weakness: Arms & Legs; Symmetric; Distal > Proximal
- Muscle atrophy: Distal legs
- Sensation: Normal
- Tendon reflexes: Brisk
- Bulbar: Normal
- Course: Slow progression; Survival up to 20 years
- Laboratory
- EMG: Denervation, diffuse; Fibrillatiions, Motor units large
- Motor neurons: Mitochondrial defects
- Serum CK: Midly high
Pontocerebellar hypoplasia + Spinal Muscular Atrophy (PCH1B)
● Exosome component 3 (EXOSC3)
- Epidemiology: 50 patients; 31% of PCH1
- Genetics
- Mutations: Missense (Most common); Deletion; Splice-site
- Common mutation: Asp132Ala; Homozygous associated with milder phenotype
- Altered reading frame mutations: More severe; Death in childhood
- No homozygous nulls reported
- Allelic disorders
- EXOSC3 protein
- RNA exosome core component
- Cellular locations: Cytoplasm; Nucleus; Nucleolus
- Exosomes
- EXOSC1–EXOSC3: Cap of exosomal ring for RNA recognition & binding
- EXOSC4–EXOSC9: Form core, a hexamer channel through which RNA passes
- Exosome-related disorders
- Cerebellar
- PCH: EXOSC1
- PCH1B: EXOSC3
- CABAC: EXOSC5
- SMA + Cerebellar hypoplasia (PCH1C) - EXOSC8
- Cerebellar atrophy + Spinal Motor Neuropathy: EXOSC9
- Spinal motor neuropathy: RBM7
- Immune: PM/Scl (EXOSC9 & EXOSC10) autoantibodies
- Cerebellar
- Clinical
- Onset: Congenital
- Motor neuron: Muscle wasting
- Contractures: Distal
- Cerebellar
- Oculomotor apraxia
- Microcephaly: Progressive
- Developmental delay: Global; Severe
- Laboratory
- MRI: Cerebellar atrophy; Brainstem & Cortex small
- EMG: Neurogenic; Large motor units
- NCV
- CMAP amplitude: Small
- Sensory responses: Normal
- Autopsy
- Cerebellum: Neuron loss (Purkinje & Granule)
- Spinal cord: Motor neuron loss
- EXOSC3 Variant syndrome: Spastic paraplegia, Early onset & Complicated
104
- Epidemiology: Bangladesh-Italian & Arab-Israel families
- Genetics
- Inheritance: Recessive
- EXOSC3 mutations: V80F; D132A; G191C
- Clinical
- Onset age: 1 to 4 years
- Spasticity
- Legs > Arms
- Gait disorder
- Tendon reflexes: Brisk
- Plantar response: Extensor
- Cognitive impairment: Mild
- Speech disorder
- Learning dificulty
- Cerebellar:
- Dysarthria
- Nystagmus
- Intention tremor & Dysmetria
- Motor: 1 family
- Distal amyotrophy
- Tongue atrophy
- Fasciculations
- Eye: Strabismus (50%)
- Skeletal: Adducted thumbs, Talipes valgus; Short stature (50%); Normal head circumference
- Course: May be progressive after 1st decade
- Laboratory
- EMG: Distal denervation
- Nerve conduction: Velocities normal
- Brain MRI: Cerebellar atrophy
- Muscle biopsy: Type 2 predominance; Varied muscle fiber size
- SSEP: Delayed central conduction times
Cerebellar atrophy + Spinal Motor Neuropathy (PCH1D)
● Exosome component 9 (EXOSC9)
- Nosology: Ponto-Cerebellar Hypoplasia 1D
- Epidemiology: 4 patients
- Genetics
- Mutations: Stop; c.481C>T, c.41T>C (Milder phenotype)
- EXOSC9 protein
- Exosome core protein
- Part of hexamer channel through which RNA passes
- PM/Scl antigen
- Other exosome disorders
- Clinical
- Onset age: Prenatal or Congenital
- Prenatal: Reduced movements; Growth retardation
- Infant: Poor head control
- Motor
- Delayed development
- Hypotonia
- Weakness: Generalized; Proximal & Distal; Respiratory
- Fasciculations: Tongue; Limbs
- Sensation: Normal
- Tendon reflexes: Reduced or Present
- Skeletal
- Joint contractures & Arthrogryposis
- Neonatal fractures
- Face: Dysmorphism
- Growth delay
- Seizures: 1 patient
- Course: Progressive
- Laboratory
- EMG: Motor neuropathy; Fasciculations
- NCV: Motor axon loss
- Muscle biopsy: Fiber type groups; Grouped atrophy; Type 1 fibers large
- Brain MRI: Cerebellar atrophy, progressive
- Serum CK: Borderline
Ponto-Cerebellar Hypoplasia + Spinal Motor Atrophy & Arthrogryposis (PCH)
● Kinesin Family Member 26B (KIF26B)
- Epidemiology: 1 Indian patient
- Genetics
- Mutation: Missense, Gly546Ser; Heterozygous
- KIF26B protein
- Kinesin, Unconventional
- Intracellular motor protein
- Cell polarity of migrating endothelial cells during angiogenesis
- Clinical
- Onset age: Congenital
- Face dysmorphism: Microcephaly
- Skeletal: Arthrogryposis; Camptodactyly; Dislocated joints
- Respiratory failure
- Course: Death at 7 months
- Laboratory
- Brain MRI: Microcephaly; Ponto-Cerebellar Atrophy; Spinal cord thin; Atrophy, progressive
- Muscle biopsy: Atrophic fibers
- Nerve biopsy: Reduced numbers of myelinated axons
- EMG: Motor neuron loss
Lower motor neuron syndrome, Childhood onset (HMNR4; DSMA4)
● Pleckstrin homology domain-containing protein, Family G, Member 5 (PLEKHG5)
- Epidemiology: Several families
- Genetics
- Mutations
- Location: C-terminal region common
- Pro630His; Pro27Ter; Val455Gly; Gln550X; Met557Leu; Phe647Se; c.2057delT, c.2752_2753delGG; Thr686Met; Pro707His
- Allelic disorders
- CMT-RIC
- DSMA4
- CMT, Axonal
- Same mutations may produce DSMA4, CMT-RIC or Overlap phenotype
- Mutations
- PLEKHG5 protein
- Cytoplasmic
- Distribution: Ubiquitous; High in peripheral nerve (endoneurium), spinal cord & brain
- Function: Nuclear Factor κB–Activator
- Mutant protein
- Reduced levels in cells
- Loss of NFκB-activating function
- Aggregates
- Small GTPase signalling pathways: Dysregulation
- Autophagy disturbed
- Proteins sharing PH or PH/RhoGEF domain: Dynamin 2; Alsin
- Other guanine nucleotide exchange factor (GEF) disorders
- Clinical
- Onset
- Age: 2 to 25 years
- Weakness
- Weakness
- Distribution: Generalized; Proximal ≥ Distal; Symmetric
- Respiratory: With disease progression
- Scapular winging
- Face & Cranial nerves (Bulbar): Normal
- Course
- Progressive over a decade to severe disability
- Milder phenotype in some patients
- Tendon reflexes: Reduced or Absent
- Contractures: Hips; Elbows; Hands
- Spine: Hyperlordosis; Scoliosis
- Intelligence: Normal
- Sensory: Normal or Mild loss
- No upper motor neuron signs
- Onset
- Laboratory
- EMG: Denervation; Motor units large
- NCV
- Motor: Normal or Slow velocities; CMAPs may become small
- Sensory: Normal or Mildly slow
- Serum CK: Mildly high
- Muscle biopsy: Denervation
- Muscle MRI: Fatty replacement in glutei & thighs > legs
- Variant syndrome: Charcot-Marie-Tooth disease, Recessive Intermediate C (CMTRIC)
- Epidemiology: Korean, Portuguese, Brazilian & Moroccan families
- PLEKHG5 Genetics
- Inheritance: Recessive
- Mutations
- Compound heterozygous or Homozygous
- Stop; Deletion; Duplication (7 bp); Missense
- Allelic with: DSMA4
- PLEKHG5 protein
- Clinical
- Onset age: 1st to 5th decades
- Weakness
- Distal
- Proximal: With disease progression; Legs & Arms
- Arms & Legs
- Course: Progressive
- Tendon reflexes: Reduced or Absent
- Sensory loss
- Distal
- Legs > Arms
- Modalities: Large & Small axon
- Varied severity
- Skeletal: Pes cavus; Spine deformity in 1 family
- Laboratory
- Serum CK: May be mildly increased
- NCV
- Motor: Intermediate slowing (24 to 39 m/s in median nerve)
- SNAPs: Absent or Reduced amplitude
- EMG: Denervation, Distal
- Muscle MRI: Fatty replacement, especially anterior & lateral distal legs
- Normal: VER; BAER
- Nerve biopsy
- Axon loss: Large > Small; Severe
- Regenerating axon clusters
- Hypomyelination
- PLEKHG5: Absent from axons; Present in Schwann cell nuclei
- Muscle: Neurogenic
Distal Motor Neuropathy with Young Adult Onset (HMNR5; DSMA5; dHMN)
● DNAJ/HSP40 Homolog, subfamily B, Member 2 (DNAJB2; HSJ1)
- Epidemiology: 5 families
- Genetics
- Mutations
- Homozygous
- Splice site (c.229+1G>A, c.352+1G>A); Deletion (c.310delC, 3.8-kb deletion); c184C>T;
- Recessive mutations: Loss-of-function
- Allelic disorders
- Other DNAJ disorders
- Mutations
- DNAJB2 (HSJ1) Protein
- Molecular Co-Chaperones
- HSP40/DNAJ family
- Cellular location: Nuclear & Cytoplasmic
- Isoforms
- 2 J-domain co-chaperones: Varied C-termini
- HSJ1b
- Membrane anchored
- Cytoplasmic face of endoplasmic reticulum (ER) by C-terminal prenylation
- Perinuclear
- Promotes proteasomal degradation of ER-located proteins: Via ER-associated degradation (ERAD) pathway
- HSJ1a
- Cytosolic or Nuclear
- Counteracts TDP-43 aggregation: Promotes HSPA-mediated refolding
- Expression: Strong in neurons; Post-synaptic NMJs in muscle
- Protects ubiquitylated clients against activity of ubiquitin hydrolases
- Mediates degradation of HSPA client proteins: Via ubiquitin–proteasome system
- J-Domain
- Stimulates ATPase activity of HSPA (Hsp70) chaperones
- Enables chaperone cycle: Alternating binding & release of client (substrate) proteins
- Has client-specific refolding activity
- May reduce aggregate formation
- Interactions: STUB1
- Mutation: Causes truncated protein
- Clinical
- Onset
- Age: 1st to 4th decade
- Foot drop
- Gait disorder
- Weakness
- Distal: Severe in legs
- Legs > Hands
- Bulbar & Respiratory: Later in course
- Course: Progressive to wheelchair
- Sensory: Normal early; Loss later in course
- Tendon reflexes: Absent
- CNS: Parkinson disease in some patients
- Edema: Legs
- Onset
- Laboratory
- Electrodiagnostic
- CMAP amplitudes: Reduced, especially in legs
- NCV: Normal velocity
- SNAPs: Normal
- EMG: Denervation, Distal legs
- Muscle: May have rimmed vacuoles
- Electrodiagnostic
- DNAJB2 (HSJ1) variant: AR-CMT 2
117
- Epidemiology: 4 families
- Genetics
- Inheritance: Recessive
- Mutations: Homozygous; ; Most stop, Some missense; Tyr5Cys, c.145delG, c.310delC, c.619-1G>A
- Clinical
- Onset ages: 2nd or 3rd decade
- Weakness
- Foot & toe dorsiflexion: Severe
- Hands & Arms
- Hip & knee flexors
- Gait: Unstable
- Sensory: Leg paresthesias
- Tendon reflexes: Absent in legs
- Some patients/families
- Parkinsonism
- Hearing loss: May be severe
- Course: Progression to severe weakness
- Laboratory
- NCV: Axonal motor & sensory length-dependent neuropathy
- EMG: Denervation, Distal legs
- Nerve pathology: Loss of myelinated axons
- Muscle CT: Fat replacement in leg & thigh muscles
- Skin: Loss of DNAJB2 in axons
- DNAJB2 variant: Neuromyopathy
195
- Epidemiology: 1 family; Son, sib & possibly mother
- Genetics
- Inheritance: Dominant
- Mutation: c.832T>G (p.*278Glyext*83)
- Mutation effects
- Abolishes stop codon on DNAJB2a isoform
- Dominant negative
- Clinical
- Onset age: 5th decade
- Gait disorder: Imbalance; Slow; Ataxia
- Weakness: Legs, distal; Fatigue
- Muscle atrophy: Distal legs
- Sensory loss: Vibration, distal legs; Joint position
- Paresthesias: Distal legs
- Tendon reflexes: Reduced or absent in legs
- CNS: Normal
- Course: Slow progression
- Laboratory
- Serum CK: 7x increased
- Head & Spine MRI: Normal
- Muscle MRI: Myopathy & Neuropathy; Fatty replacement
- NCV: Axon loss, sensory & motor; Long F-wave & distal latencies; Velocities normal
- EMG: Chronic neurogenic; No spontaneous activity
- Muscle pathology: Fiber type groups; Grouped atrophy; Internal nuclei; Vacuoles
- Nerve pathology: Axon loss
Lower motor neuron syndrome with late-adult onset (SMAJ; LOSMoN)
● CHCHD10
- Nosology: Spinal muscular atrophy, Jokela type
- Epidemiology
- 17 Finnish families
- Eastern Finland (Northern Karelia) prevalence: 12:100,000
- Genetics
- Mutation: G66V
- Allelic disorders
- CHCHD10 protein: Mitochondrial
- Clinical
- Onset
- Age: 30 to 73 years
- Weakness, Cramps or Fasciculations
- Weakness
- Proximal > Distal + Abdomen
- May be: Asymmetric or Distal
- Cramps: Proximal > Distal; Painful
- Fasciculations: Distal & Proximal
- Tendon reflexes: Absent diffusely or in legs
- Other occasional features
- Myalgias (50%)
- Drop attacks: Body or head
- Tremor: Associated with hand fasciculations
- Ataxia
- Pes cavus: Mild
- Vibration sense reduction (50%): Distal legs; Asymmetric
- Course
- Progression: Slow
- Most retain some ambulation
- Onset
- Laboratory
- Serum CK: Normal to 8x High
- NCV
- Normal velocities
- CMAP amplitudes: Normal or Reduced
- SNAPs: Normal or Mildly small
- EMG: Denervation, Distal & Proximal
- Motor unit potentials: Long duration; Large amplitude
- Fibrillations
- Fasciculations
- Complex repetitive discharges
- Recruitment: Reduced
- MRI
- Posterior lower leg muscles abnormal
- Especially medial gastrocnemius
- Muscle biopsy
128
- Fiber type grouping
- Grouped atrophy
- Pyknotic nuclear clumps
- Type 2 fibers: Small
- Largest muscle fibers: Hypertrophy
- Neonatal myosin heavy chain (MHCn): 3%-50% (median 15%) of muscle fibers
- Vacuoles
- Frequency: 60% of biopsies
- Fibers also stain for LC3, p62, SMI-31 & TDP-43
- More common with longer disease duration
Proximal Spinal Muscular Atrophy with Progressive Myoclonic Epilepsy (SMA-PME; SMAPME)
● N-Acylsphingosine Amidohydrolase 1 (ASAH1; Acid ceramidase; Acylsphingosine deacylase)
- Epidemiology: 50 patients
- Genetics
- Mutations
- Types: Missense (Thr42Met (Common), Thr42 Ala (Milder phenotype), Lys152Asn (Deafness)); Deletion, Small or whole gene
- Homozygous
- Allelic disorders
- Farber Lipogranulomatosis: Very low ASASH1 levels
- Progressive adult-onset brachydactyly due to osteolysis
- SMA without epilepsy
- Mutations
- ASAH1 protein
- Localization: Lysosome
- Maintains intralysosomal ceramide homeostasis
- Cleaves Ceramide into Sphingosine & free fatty acids at acid pH

- Clinical
- Onset age: 1 to 30 years; Mean 6 to 10 years
- Weakness
- Proximal
- Legs, then Arms; Face, mild
- Respiratory: Later in disease course
- Dysphagia
- Tongue fasciculations
- Tendon reflexes: Absent or Present
- Tremor: Postural
- Seizures
- Myoclonus epilepsy
- Drop attacks
- With more severe phenotypes
- Learning dificulties (50%)
- Hearing loss
- No contractures
- Skin: Normal
- Course
- Progressive: To severe disability
- Death: Some patients in 2nd decade
- Laboratory
- EMG: Chronic denervation
- NCV: CMAPS small; SNAPs normal
- EEG
- Epilepsy: Subcortical myoclonic epileptiform activity, sensitive to hyperventilation
- May be normal
- Serum CK: Normal
- Muscle biopsy: Denervation & Reinnervation
- Skin biopsy: Zebra bodies (50%); Farber, Banana bodies unusual
- Brain MRI: Often normal
- Variant syndrome: Farber Lipogranulomatosis
- Genetics
- ASAH1 mutations: Cause severe reduction in Acid ceramidase activity (< 10%)
- Inheritance: Recessive
- Clinical
- Skin: Lipogranulomata, subcutaneous; Periarticular subcutaneous nodules
- Joints: Contractures; Pains
- Hoarse voice
- CNS: Mental retardation; Progressive deterioration
- Lower motor neuron involvement (15%)
- Hypotonia
- Muscle atrophy
- Respiratory insufficiency
- EMG: Denervation
- Systemic
- Hepatomegaly
- Splenomegaly
- Eyes: Macular cherry red spots
- Death: < 2 years
- Laboratory
- NCV: Slowing in some patients
- Nerve pathology
- Schwann cells, myelinating: Banana bodies
- Genetics
Spinal Muscular Atrophy with Hypomyelination & Cerebellar Hypoplasia (PCH1C)
● Exosome component 8 (EXOSC8)
- Epidemiology: 3 families & 22 patients, Hungarian & Arab-Palestinian
- Genetics
- Mutations: Missense; Ala2Val, Ser272Thr
- Other exosome disorder: PCH1B - EXOSC3
- EXOSC8 protein
- Exosome component
- Multi-protein complex
- Functions
- 3'->5' exoribonuclease activity
- Participates in many cellular RNA processing & degradation events
- Degradation of AU-rich element (ARE) containing mRNAs
- Immune disorders: Exosome = PM/Scl target of autoantibodies
- EXOSC8
- Part of central hexamer channel of exome
- Non-catalytic component
- Cell location: Nucleus & Cytoplasm
- Exosome component
- Clinical
- Onset age: Infant; 2 to 4 months
- Failure to thrive
- Weakness: Severe
- Spasticity
- Psychomotor retardation
- Vision: Impaired
- Hearing: Impaired
- Deterioration: Triggered by inter-current infections
- Death: Respiratory failure < 20 months
- Laboratory
- CNS
- Cerebellar hypoplasia
- Hypomyelination: Especially lateral descending tracts
- Muscle
- Fiber size: Varied
- Cytochrome oxidase: Some negative fibers
- Mitochondrial oxidative enzymes: Complex I & IV reduced
- Peripheral nerve: Normal myelin
Encephalopathy with Distal Spinal Muscular Atrophy, Early-onset, Progressive (PEAMO)
● Tubulin-specific chaperone E (TBCE)
- Epidemiology: > 15 patients
- Genetics
- Mutations: c.463Tgt;A (p.Ile155Asn) homozygous or + Stop
- Allelic disorders
- Sanjad-Sakati syndrome (HRDS)
- Most common TBCE syndrome
- Mid-East origin common
- Genetics: c.155_166del, homozygous mutations common
- Clinical features
- Congenital hypoparathyroidism
- Mental retardation
- Face dysmorphism
- Growth failure
- Kenny-Caffey syndrome 1
: HRDS + Osteosclerosis & Infections
- Myopathy-Neurodevelopmental
- Spastic paraparesis, complex
- Familial hypoparathyroidism
- Lethal: Complete loss of TBCE function
- Sanjad-Sakati syndrome (HRDS)
- TBCE protein function
- Expression: Ubiquitous
- Tubulin-specific chaperones
- TBCA-TBCE: Process tubulin chains
- After nascent α- & β-tubulin chains are folded by cytosolic chaperonin (CCT/TRiC) complex
- TBCE: Interacts with α-tubulin; Folding; Prevents degradation
- TBCE + TBCD: Formation & disassembly of α/β-tubulin heterodimers
- Heterodimer polymerization into microtubules
- See: SMA +
- Clinical
- Onset age: Neonatal to 25 years
- Hypotonia
- Developmental delay
- Speech: Absent or Dysarthria
- Cognition: Moderate to Severe involvement
- Weakness & Wasting
- Distal
- Legs & Arms
- Spasticity: Tetraparesis
- Ataxia (60%)
- Optic atrophy (40% to 90%)
- Strabismus/Gaze palsy
- Seizures
- Scoliosis
- Course: Progressive
- Laboratory
- Brain MRI
- Cerebellar atrophy
- Corpus callosum hypoplastic/thin
- Similar to NBIA
- Muscle pathology: Small angular fibers; Fiber type grouping
- EMG: Motor units large; Distal fibrillations; No fasciculations
- NCV: CMAP amplitudes reduced; Conduction velocities normal
- VEPs & BAERs: Delayed
- Calcium/Phosphate metabolism: Normal
- Brain MRI
- TBCE variant: Myopathy-Neurodevelopmental
229
- Epidemiology: Eastern Europe; 10 patients
- Genetics
- Mutation: c.100+1G>A
- Clinical
- General: Less severe than other TBCE syndromes
- Onset: Childhood; 1 month to 18 years
- Myopathy: Weakness & Amyotrophy
- Axial & Proximal muscles (90%)
- Nocternal hypoxemia
- Developmental delay/Mild intellectual disability (100%): Delayed language
- Skeletal: Scoliosis (60%); Rigid spine; Growth delay/Short stature
- Endocrine: Testicular failure (70%); Cryptorchidism; Hypergonadotropic hypogonadism
- Growth failure: Growth hormone deficiency (30%)
- Laboratory
- Eosinophilia
- Neutropenia
- Muscle MRI: Involvement of Gluteus maximus, Vastus lateralis, Semitendinosus
- EMG: Myopathic or Normal
- Muscle pathology
- Non-specific myopathic changes
- Ultrastructure
- Actomyosin cytoskeleton degradation
- Nuclei: Detachment of heterochromatin from nuclear envelope
- Brain MRI: Normal or Pituitary hypoplasia, Corpus callosum hypoplasia
- Mouse model: pmn
- Genetics
- TBCE Mutation: Trp524Gly
- Gene location: Mouse chromosome 13
- Loss of motor neurons & myelinated axons
- CNS
- Tracts - Fasciculus gracilis; Rubrospinal; Reticulospinal
- Normal: Corticospinal tract
- Pathological pattern: Dying back
- Death: 6 weeks
- Genetics
Spinal Muscular Atrophy, Early-onset with Neurodegeneration 136
● Tubulin-specific chaperone D (TBCD)
- Epidemiology: 1 family 2 patients
- Genetics
- Mutation: Homozygous; R942Q
- Allelic disorder
- Encephalopathy, progressive, early-onset, with Brain atrophy & Thin corpus callosum
: Has motor neuron loss
- Encephalopathy, progressive, early-onset, with Brain atrophy & Thin corpus callosum
- TBCD protein
- Microtubule assembly
- Peripheral nerve: Axon & Dendrite maintenance
- See: dHMN + TBCE mutations
- Clinical
- Hypotonia
- Developmental disorder
- Weakness: Diffuse; Prfoximal > Distal; Face; Respiratory
- Fasciculations: Tongue
- Microcephaly
- Seizures
- Psychomotor retardation
- Vision loss: Optic atrophy
- Laboratory
- Serum CK: Normal
- Brain MRI: Cerebral atrophy
- NCV: CMAP amplitudes reduced; Velocities reduced
- Muscle: Grouped atrophy; Hypertrophy of largets muscle fibers
- Sural nerve: Normal
Motor Neuropathy & Intellectual Disability (TBCK Encephaloneuronopathy; IHPRF3)
● TBC1 Domain-containing Kinase K (TBCK)
- Epidemiology
- > 10 patients
- Common: Puerto Rican children of Boricua descent (Arg126X); Carrier rate 1:45
- Genetics
- Mutations: c.1652T>C (p.L551P); Arg126X
- Allelic disorder
- Hypotonia, infantile, with Psychomotor retardation & Characteristic facies 3
: > 100 patients; mtDNA reduced
- Heterozygotes: Deformities, Toe & Foot; Apraxia; Peripheral neuropathy
- Hypotonia, infantile, with Psychomotor retardation & Characteristic facies 3
- TBCK protein
- Protein kinase
- Associates
- Mitotic apparatus
- Proteins: PPP1R21; c12orf4; CRYZL1
- Vescicular organelles in neurons
- Ferry complex: Transport mRNAs
- Regulates: Cell size, Cell proliferation, MTOR
signaling
- Axonal mRNA trafficking
- Clinical
- Onset age: Child
- Hypotonia
- Weakness
- Diffuse: Distal > Proximal
- Respiratory insufficiency by teenage years
- Rarely sit
- Tendon reflexes: Absent
- Face
- Ptosis
- Hypotelorism
- Coarse
- Macroglossia
- CNS
- Autism
- Speech & Cognitive delay
- Seizures: Focal & Generalized
- ? Exacerbation by ketogenic diet
- Course: Progressive
- Systemic
- Osteoporosis
- Renal: Calculi; Urinary retention
- Hypothermia: Intermittent to 33°C
- Brachymelia
- Course: Progressive
- Laboratory
- Motor axon loss
- Dyslipidemia: HIgh cholesterol or triglycerides
- TBCK- fibroblasts: Increased LC3+ autophagosomes
- Urine: Free oligosaccharide profiles
- Brain MRI: Leukoencephalopathy; Atrophy
- NCV: Motor neuronopathy
- CMAPs: Small amplitude
- SNAPs: Generally preserved
- Velocities: Normal
- EMG: Chronic partial denervation; Myokymia in 1 patient; Some myopsthy in teenage years
- Muscle ultrasound: Atrophic; Dense; Fasciculating movements
- Muscle & Nerve biopsies: Non-diagnostic; COX deficiency
MTDPS18: Spinal Muscular Atrophy-like disorder
● Solute carrier family 25 (Mitochondrial oxodicarboxylate carrier), Member 21 (SLC25A21)
- Epidemiology: 1 Pakistani patient
- Genetics
- Mutation: Lys232Arg; Homozygous
- SLC25A21 protein
- Mitochondrial
- Oxodicarboxylate carrier
- Clinical
- Onset age: 3 years
- Weakness
- Distal then Proximal
- Arms & Legs
- Face: Mild
- Respiratory
- Gait disorder
- Muscle atrophy
- Exacerbations: Associated with fever
- Tendon reflexes: Brisk
- Scoliosis
- Course: Progressive; Wheelchair at 15 years
- Laboratory
- Anemia: Microcytic
- NCV: Motor axon loss
- Nerve: Axon loss
- Muscle
- Groups of
- Atrophic & Hypertrophied muscle fibers
- Fiber types
- Type 1 predominance
- Mitochondrial
- COX- muscle fibers
- Complex I & IV activities: Reduced
- mtDNA depletion
- Groups of
- Brain MRI: Normal
- CSF: Lactate mildly high
- Urine: Excretion of 3-Hydroxyisovaleric & Glutaric acid
Motor Neuropathy, Distal 185
● Neuronal cell adhesion molecule (NRCAM)
- Epidemiology: 2 families
- Genetics
- Mutations: Ser134Pro; Gln25X
- Allelic disorder: Neurodevelopmental disorder with Developmental delay, Hypotonia, Spasticity
- NRCAM protein
- Location: Neurons & Glial cells in Ventral midline
- Adhesion molecule
- Immunoglobulin (Ig)-CAM
- L1 subgroup
- Neural specific
- Nervous system development
- Axon pathfinding
- Clinical
- Onset age: 2nd decade
- Weakness
- Distal
- Legs > Arms
- Skeletal: Scoliosis; Pes cavus; Hammer toes
- Laboratory
- EMG: Denervation, distal; Myopathic, proximal
- NCV: CMAPs small; SNAPs normal
- Serum CK: 2x to 3x high
- NRCAM variant: Neurodevelopmental disorder with Developmental delay, Hypotonia, Spasticity (NEDNMS)
- Epidemiology: 8 patients
- Genetics
- Mutations: Missense & Stop
- Inheritance: Recessive
- Clinical
- Onset age: Birth
- Developmental delay
- Cognitive disorder
- Hypotonia (50%)
- Spasticity (50%)
- Ataxia
- Skeletal: Dysmorphism; Microcephaly; Scoliosis; Hip dislocation
- Cryptorchidism
- Respiratory: O2 supplementation
- Eye: Strabisums (30%); Optic atrophy
- Hearing loss
- Laboratory
- Brain imaging: Ventricles large; Myelination Δ
- NCV: Polyneuropathy
Motor Neuropathy 194
● Barrier-to-Autointegration Factor 1 (BANF1)
- Epidemiology: 1 patient
- Genetics
- Mutation: Missense; Gly16Arg
- Allelic disorder: Néstor–Guillermo progeria
: Ala12Thr mutation
- BANF1 protein
- Locations: Nuclear lamina; Phosphorylated forms in cytoplasm
- Regulation of gene expression, cell cycle progression, nuclear integrity
- DNA binding: Bridges double-stranded DNA into ordered nucleoprotein complex
- Loss: Absence of mitotic cells; Aberrant nuclear lamina structure
- Interacts with: Histones; Chromatin modifying proteins; Lamin A/C; Emerin; PARP1 in response to oxidative stress
- Clinical
- Onset age: 3 years
- Gait disorder
- Weakness
- Diffuse: Complete foot drop
- Legs > Arms
- Respiratory
- Tendon reflexes: Reduced at ankles
- Sensation: Cold reduced at toes
- Intention tremor: Mild
- Course: Slow progression
- Laboratory
- NCV: Normal velocities
- EMG: Denervation, diffuse
Motor Neuropathy + Tremor & Febrile Seizures 212
● Phosphatidylinositol Glycan Anchor Biosynthesis Class G Protein (PIGG)
- Epidemiology: 9 patients, 7 families
- Genetics
- Mutations: Stop, Splice, Missense
- Allelic disorder
- Neurodevelopmental disorder ± Hypotonia, Seizures & Cerebellar atrophy (NEDHSCA)
- Blood group, EMM System (EMM)
- Neurodevelopmental disorder ± Hypotonia, Seizures & Cerebellar atrophy (NEDHSCA)
- PIGG protein
- Endoplasmic reticulum
- Biosynthesis of glycosylphosphatidylinositol (GPI)
- Transfer of ethanolamine phosphate to 2nd mannose of GPI H7 to form H8
- GPI-anchored proteins: Contactin-1 & Contactin-2
- PIGG disorders: May disrupt node of Ranvier
- Clinical
- Onset age: 4 months to Teens
- Motor neuropathy
- Distal
- Legs
- Minimally progressive
- Falls
- Muscle activity after exertion
- Foot deformity
- Tremor: Postural
- Febrile seizures: Resolve < 6 years
- Ataxia: Mild
- Laboratory
- NCV: Motor neuropathy; Conduction block; Temporal dispersion
- EMG: Myokymia; Nerve hyperexcitability
- MRI
- Nerves: Thick or Normal
- Muscle: Symmetric fat replacement of Gastrocnemius & Soleus; Thigh normal
CMT 2KK: Motor-Predominant Neuropathy
● RHO GTPase-Activating Protein 19 (ARHGAP19)
- Epidemiology: 25 patients; 20 families
- Genetics
- Mutations: Loss of function
- ARHGAP19 protein
- Cell location: Nucleus
- Negative regulators of Rho GTPases
- Involved in cell migration, proliferation, differentiation, actin remodeling, G1 cell cycle progression
- Clinical
- Onset age: Mean 10 years; Range infancy to 20 years
- Weakness (90%)
- Distal
- Legs > Arms (90%)
- Asymmetry
- Sensory loss (64%)
- Tendon reflexes: Reduced
- Skeletal: Pes cavus
- Course
- Slow progression
- Some with rapid onset
- Laboratory
- NCV: Motor axon loss; Some with slowed velocities or conduction block
SMA: Severe infantile
● Recessive
Scapuloperoneal syndromes (? Dominant; Sporadic)
ALS Genes: Hereditary
& Susceptibility
|
|
Protein Pathways Protein Quality SOD1 VCP OPTN TBK1 UBQLN2 p62 NEK1 FIG4 CCNF CYLD Cytoskeletal PFN1 DCTN1 TUBA4A EPHA4 KIF5A ANXA11 PRPH RNA-related ANG ARPP21 ATXN2 c9orf72 ELP3 FUS NEK1 TAF15 TDP43 TIA1 Vesicles Alsin CHMP2B VAPB Lipids SPTLC1 SPTLC2 |
General: Hereditary vs Sporadic ALS
| Feature | Hereditary ALS | Sporadic ALS |
| Males:Females | 1:1 | 1.7:1 |
| Disease Duration | Bimodal < 2 & > 5 years |
Unimodal 3 to 4 years |
| % of ALS cases | 5% to 15% | ~90% |
| Onset | ||
| Age distribution | More younger | More older |
| Mean age | 46 years | 56 to 63 years |
| Juvenile | ALS 2, 4, 5, 6 | Rare |
| Bulbar features | 25% c9orf72 40% ALS1 10% |
15% |
| Legs | Common | Occasional |
| LMN predominant | ALS 1, 6, 17 | PMA |
ALS SYNDROMES: DOMINANT
- Amyotrophic Lateral Sclerosis 1 (ALS 1)
● Superoxide Dismutase 1 (SOD1); Chromosome 21q22.11; Usually Dominant
Clinical features
General
Specific syndromes
Mutations
Location
Functional aspects
Specific correlations
Pathology
Population statistics
SOD1 other
SOD1 protein
Variants
Canine disorder
SOD1 deficiency
From: M WeissALS-SOD1 (A4V): Tongue atrophy Early description of SOD1 ALS family with A4V mutation
Early description of SOD1 ALS family with A4V mutation
- Population statistics
- Frequency of SOD mutations in ALS syndromes
- 12% to 23% of Familial ALS
- 1% to 3% of Sporadic ALS
- 1% to 3% of all ALS
- China: Most common F-ALS gene (59%)
- Penetrance: Overall 85% by age 85
- Frequency of SOD mutations in ALS syndromes
- Genetics
- Inheritance: Dominant; de novo; Recessive
- Mutations: Location & Character
- > 200 different disease related mutations identified
- 55 likely pathogenic: Based on linkage, population studies, or hotspot location
- Locations
- Usually in exons 1, 2, 4 & 5
- Few in exon 3: 6 exon 3 mutations identified
- 6 different mutations identified at codon 93 (Gly93) in exon 4
- Most common mutations: D90A; A4V; I113T; L144F; G41D; E100K
- Hot spots: A4; C6; N86; G93; D101; L126; N139;L144
- Mutations: Common features
- Heterozygous
- Missense
- Point
- Dominant inheritance with complete penetrance
-
A4V; G37R; L38V; G41S; H43R; H46R; D76V; L84F; L84V;
N86K; E100G; D101F; I104F; G108V; C111Y; I112M; G114A;
L126X; G127X; G141E; L144F; V148G; V148I - China 181: V47A, H46R, C111Y, G147D
- Finland: D90A
- Scotland: Ile114Thr
- Dominant de novo
179
- Slow progression: Gly38Arg; His47Arg
- Other: His81Arg; Ala90Thr; Asp91Val; Asp102Asn (Rapid progression)
- Other types
- Stop codon
183
- 17 nonsense mutations identified
- Nonsense mutations producing disease
- Locations: Outside AA 50 to 100
- Escape nonsense mediated decay
- Produce polypeptide length of 121 tp 156 aa (Normal 153)
- Exon 5 not needed for toxic action of mutated SOD
- Intron: IVS4AS, A-G, -11 (11 bases upstream from intron-junction of exon 5)
- Deletion
- Intronic: Altered splice site; 3' untranslated region
- Recessive
- D90A; ? D96N
- Dose effect (Homozygous more severe): L84F, N86S, L126S
- Incomplete penetrance 76
- Stop codon
183
- External link: ALS mutation database
- Mutations: Functional aspects
- Most mutations affect subunit protein folding or dimer contact.
- Mutations may alter some Cu binding sites
- Mutant SOD enzyme activities vary.
- Inactive: H46R & Gly85Arg
- Normal: C6S; E40G; L84F; Asp90Ala; G93A & G93D; D109Y
- Protein stability
- Unstable: L126X; G127X
- Normally stable: A89V; D90A
- Correlation between ratio of mutant:normal SOD1 in RBC
53
- Lower ratio: Shorter disease duration
- No correlation between duration of disease and SOD mutant peptide ...
- Ability to scavenge superoxide
- Half-life
- Solubility
- Resistance to proteolytic digestion
- Propensity to aggregate spontaneously into sedimentable structures
- Relative affinity for Cu
- Stop mutations all in exons 4 & 5: Active site in exon 3 preserved
- Allelic disorders
- SOD1 deficiency
- Motor predominant neuropathy, Dominant
- Animal variant: Myelopathy
- SOD1 protein
- Abundant: 1% of cytosolic protein
- Structure: Homodimer
- Each subunit contains Cu++ & Zn++ in cave-like active site
- Subcellular location: Cytoplasmic & mitochondrial
- Function
- Detoxifies O2- to H2O2
- Prevents oxidative damage
- Associated enzymes preventing oxidative damage: Catalase; Glutathione peroxidase
- Zn++ maintains pH stability of dismutase reaction
- Misfolded SOD1: Binds to VDAC1 78
- Other SOD molecules
- SOD2: Mitochondrial; Manganese
- SOD3: Extracellular; Copper-Zinc
- Clinical: General features of hereditary SOD1 ALS
- Clinical vs Sporadic ALS
- Fewer: Upper motor neuron signs
- Onset with monomelic leg weakness: Suggests familial ALS
- Most mutations with variable clinical features among patients
- Homozygosity
- Often causes more severe phenotype than heterozygous state: Asn86Ser; Asp90Ala
- Patients often have mainly lower motor neuron signs
- Onset
- Mean age
- General: 46 years
- US: 50 years
- China: 44 to 46 years
- 10 years younger than sporadic ALS
- 3.5 years younger than non-SOD FALS
- 90% penetrance by age 70
- General: 46 years
- Juvenile (< 25 years) onset: Few patients
230
- Heterozygous
- SOD1 Mutations: Gly16Ser, Gly38Arg, Pro66Ser, His80Ala, Leu84Phe, Asp125Gly
- Inheritance: de novo or Dominant with varied expression
- Onset age: 15 to 24 years
- Onset region: Commonly lower extremity
- Course: Rapid progression
- Homozygous
- Loss of function variants
- Onset: Infant
- Heterozygous
- No anticipation
- Preparetic phase: Some with myalgias, leg cramps & painful paresthesias
- Weakness: Asymmetric limb
- Mean age
- Duration
- Mean for all FALS = 3.4 to 5.6 yrs
- Similar for SOD & non-SOD FALS
- Survival
- Male > Female
- 5 years: 31% to 55%
- Varies strongly with specific mutations
- Lower motor neuron signs
- Common
- Especially predominant in rapidly progressive cases
- Upper motor neuron signs
- May be more common with slowly progressive course
- Reported with Asp76Tyr & Homozygous Asp90Ala mutations
- Dominant upper motor neuron signs: Not reported
- Bulbar onset
- Unusual
- Later onset age
- Reported with some mutations
- Cramps: G12R; G37R; D90A; E100G
- Other Neural features
- Sensory neuropathy: A89V; D90A
- Extra-motor system symptoms: ? More frequent
- Dementia: Rare; A4T; G41S; I113T; L144F; ? More common in non-SOD group
- Autonomic failure: D90A; Val118Leu; 1 patient 3
- Vocal cord dysfunction: A4V; I113F; Gly147Ser; I149T
- Supranuclear EOM paresis: May occur late in disease course; H80R; I104F
- Bladder dysfunction: G41S; Asp90Ala (Homozygous)
- Treatment
- SOD1 Antisense oligonucleotide (Tofersen), Intrathecal
184: Outcomes
- 6 months: Neurofilament Light levels reduced in serum
- 2 years: Mildly improved strength in 50%; Slow progression of weakness in 25%
- Side effects: Myelitis or Motor radiculitis; Reduced strength over weeks to months
- CSF: Increased protein & cells may have no clinical correlate
- SOD1 protein in motor neurons: Reduced 45% to 84% 231
- SOD1 Antisense oligonucleotide (Tofersen), Intrathecal
184: Outcomes
- Clinical vs Sporadic ALS
- Population statistics
| ALS syndromes: Correlations with some SOD1 mutations | |
|---|---|
|
Specific SOD1 mutations Exon 1; Ala4Val Most common mutation Rapid onset & progression (1.0 yrs) Frequently only lower motor neuron signs Exon 2; His46Arg @ Cu binding site of SOD Onset: Late; Legs Bulbar unusual Slow progression (17 yrs) Exon 2; 6 bp deletion(ΔG27/P28) 65 Mutation reduces transcription Low levels of mutant SOD1 protein Philipino founder Low penetrance Disease duration: 4.3 years Exon 4; Leu84Val Lower motor neuron only Rapid progression (1.5 yrs) ?Earlier onset in males Exon 4; Asp90Ala Onset: 20 to 94 yrs; Legs; Preparetic phase Leg cramps; Myalgia; Painful paresthesia Bladder dysfunction Progression: Slow; Legs ® Arms Inheritance Recessive: Finnish (2.5% carriers) Dominant: Clinically variable Incomplete penetrance Exon 4; Ile104Phe Variable intrafamilial clinical features Age of Onset: 6 yrs - asymptomatic Course: 2 to 14 yrs until bulbar signs Limb onset: arms or legs Exon 4; Ile113Thr Reported in Sporadic ALS patients Relatively common; Low penetrance Late Onset: Mean 59 years Course: Variable; 2 to 20 years Exon 5; Codon 126 2 base pair deletion Rapid Progression |
ALS clinical features Lower motor neuron predominant A4V; G72C; Leu84Val; Gly93Cys; E100K; D101N; S134N Slow progression C6S; Asp11Tyr; G12R; V31A; Gly37Arg (18 yrs); Gly41Asp (11 yrs); F45C; H46R131; D76V; Gly93Cys (13 yrs); Gly93Ser; Leu144Ser; Leu144Phe (9 yrs) Rapid progression Ala4Thr (1.5 yrs); A4V; C6G; C6F V7E; L8Q; G10V; G41S; H43R; H48Q Asn86Ser Homozygous (5 mo); D101>G,H,N,Y; Leu106Val (1.2 yrs); I112T; I113F; Arg115; D125H; 126 2bp del; S134N; Gly147Ser; Val148Gly (2 yrs); V148G Late onset His46Arg; Gly85Arg (55 yrs); D90A; I113T; Ala140Gly; Leu144Phe Early onset Gly37Arg; Leu38Val; A89V; L104F (6 yrs); ?Leu106Val More common in females Gly41Asp Bulbar onset Cys6Gly; L8Q; His48Gln; Asp76Tyr; Asp90Ala (Homozygous); D101Y; I112M; T116R; Cys146Arg; Gly147Ser; Val148Ile; I149T; Ile151Thr Onset in legs G10V; H46R; L84F; D90A; Gly93Cys; Gly93Ser SOD Mutations in "sporadic" ALS Most common: Asp90Ala; Ile113Thr Other: Asp11Tyr; V14G; G16S; E21K; G72S; D101N; V118InsAAAAC; E133delGAA Incomplete penetrance |
- Genetic variants: Other features
- Ala4Val
147
- Epidemiology
- Common among North American SOD1 ALS patients: 36% to 50%
- Unusual in China
- SOD1 protein: Folding of mutated SOD1 protein less stable than normal 30
- Clinical
- Onset age: Mean 50 years
- Course
- Rapidly progressive
- Mean survival: 1.4 years; 50% of other SOD1 mutations
- Lower motor neuron predominant
- Epidemiology
- Phe20Cys
60
- Onset
- Legs
- Weakness or Fasciculations
- Later in Females
- Course: Mean 1.9 years
- Bulbar signs present late
- Onset
- Gly85Arg (Transgenic mouse pathology)
- Rapid clinical course; Normal SOD1 activity
- Astrocytic inclusions: Contain SOD1 & ubiquitin; Increased with disease progression
- Aggregates in motor neurons: Contain SOD1
- Reduced Glial glutamate transporter
with disease progression
- ? 1° disease in astrocytes
- Asp90Ala
- Finnish population
- Other geographic locales
- Dominant inheritance
- Progression in heterozygotes: Typical ALS course
- SOD1 activity: Normal
- Rat SOD models
18
- G93A high expression
- Onset: 110 days
- Rapid disease progression (8 days)
- Motor neuron degeneration
- Vacuolar pathology
- H46R-4 high expression
- Onset: 140 days
- Slower progression (24 days)
- Motor neuron pathology
- Protein deposition & aggregation
- G93A high expression
- Other transgenic mice
- Mitochondrial vacuoles, or
- Neurofilament accumulation
- Mitochondrial vacuoles, or
- Ala4Val
147
- SOD1 MND Laboratory
- CSF protein: Often high, atypical for other ALS
- Pathology of SOD1-ALS: May vary with different mutations
- Common features: Some different from sporadic ALS
- Cell loss: Corticospinal tract neurons & Anterior horn
- Relatively normal corticospinal tract & brainstem motor nuclei: Gly93Cys
- Posterior column changes
- Especially fasciculus cuneatus (Middle root zone) at cervical level
- Mutations: A4V; A4T; Gly93Cys; Gly93Ser; I113T; Not H48Q
- Spinocerebellar tract pallor
- Mouse models
- Activation of non-neuronal cell types: Astroglia & Microglia
- Sequential activation of Caspase-1 and -3
- Cell loss: Corticospinal tract neurons & Anterior horn
- Hyaline conglomerate inclusions (HCI)
27
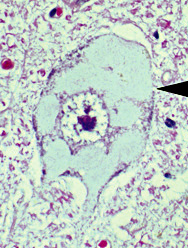
Hyaline conglomerate
inclusion- Cells: Surviving motor neurons
- Subcellular location: Intracytoplasmic
- Contain SOD, neurofilaments, peripherin ± ubiquitin
- No correlation between frequency of aggregates & cell death
- Mutations: Ala4Val, A4T, H48Q, D101N, I112T, I113T, 126 bp deletion
- Have specificity for SOD1 FALS mutations
- E100G: ALS-dementia type
- Dentate granule inclusions
- Frontal lobe gliosis
- Muscle
- Common features: Some different from sporadic ALS
- Animal variant: Canine Degenerative Myelopathy, Recessive SOD1
92
- Dog breeds
- Multiple: > 60
- Pembroke Welsh Corgi, Boxer, Rhodesian Ridgeback, German Shepherd, Markiesje, Chesapeake Bay Retriever
- SOD1 mutations
- Common: Glu40Lys (c.118G>A)
- Homozygous
- Missense
- Ancestral: Present in many dog breeds
- Penetrance
- Incomplete
- Wire Fox Terrier: High frequency of E40K allele; Low frequency of disease
- Other: Thr18Ser in Bernese Mountain Dog
- Common: Glu40Lys (c.118G>A)
- Clinical
- Onset age: 5 to 8 years
- Spastic paraparesis
- Gait ataxia
- Flaccid tetraparesis: Late
- Dyskinesia
- Course: Progressive over 2 to 3 years
- Pathology: Spinal cord
- Lateral white matter: Myelin & axon loss
- Neuronal cytoplasmic inclusions: Contain SOD1
- Early: Thoracolumbar spinal cord
- No motor neuron cell body loss
- Dog breeds
- SOD1 variant: SOD1 deficiency
155
- Nosology: Spastic Tetraplegia & Axial Hypotonia, Progressive (STAHP)
- Epidemiology: 16 patients
- Genetics
- Mutation: Homozygous; Truncating; Common c.335dupG
- Inheritance: Recessive
- Heterozygous carriers: No phenotype
- Clinical
- Onset age: Infancy
- Developmental delay: Cognition impaired
- Dysmorphism: Low set ears; Overlapping toes
- Spasticity: Tetraparesis; Legs & Arms; Tendon reflexes brisk
- Axial hypotonia
- Hyperekplexia
- Laboratory
- Brain MRI: Atrophy Cerebellar vermis, Brainstem, Frontotemporal
- Blood manganese: Low
- SOD1 activity: Undetectable
- Serum CK: Normal to 870
- Muscle
- Fiber sizes: Varied
- α-dystroglycan: Focally reduced
- Muscle ultrasound & EMG: Fasciculations
- NCV: Normal
- SOD1 variant: SOD1 Motor-predominant polyneuropathy
220
- Epidemiology: 17 patients in inherited polyneuropathy cohort
- Genetics
- Clinical
- Onset age: Mean 47 years; Range 30 to 75 years
- Weakness
- Legs (100%): Distal ≥ Proximal
- Arms (50%): Distal ≥ Proximal
- Asymmetric (75%)
- Bulbar: Normal
- Respiratory: No symptoms
- Muscle atrophy (85%)
- Fasciculations or Cramps (50%)
- Tendon reflexes: Reduced in legs (80%); Brisk in arms (25%)
- Sensory loss (50%): Mild
- Bulbar: Normal
- Course: Mean 7 years; Range 1 to 40 years
- Laboratory
- Serum CK: Normal to Mildly high
- NCV
- Motor axon loss
- Conduction velocity normal
- Sural SNAPs: Normal (75%); Reduced (25%)
- EMG: Chronic denervation
- ALS 15
82
● Ubiquilin 2 (UBQLN2; PLIC2; CHAP1);
Chromosome Xp11.21; Dominant or Semi-Dominant
- Epidemiology
- Frequency: > 10 families; ~1% of ALS
- Male vs Female
- Progression: Similar disease duration
- Onset age: Males younger; 34 vs 47 years
- Genetics
- Mutations
- Missense: Often in Proline
- T487I, Pro497His, Pro497Ser, P497L, P506T, P506S, P509S, P525S
- In or near PXX (Proline/Glycine repeats) domain
- Gene subjected to X-inactivation
- Penetrance: 90% by age 71
- Variant syndrome: Spastic paraparesis
- Female carriers: Usually asymptomatic
- Allelic disorders
- Sporadic ALS patients: Outside PXX domain
- PSP: S222G mutation
- Madras Motor neuron disease: M392L
- Hereditary spastic paraplegia: P506S
- Mutations
- UBQLN2 protein
- Ubiquitin-like protein family: Bind to subunits of proteasome
- Regulates: Protein degradation pathways
- Ubiquitin-proteasome system (UPS)
- Endoplasmic reticulum–associated protein degradation pathway: Interacts with UBXD8 & HERPUD1
- Macroautophagy
- Interacts with: TDP-43; HSP70 (HSPA1A)
- Mutations: May impair autophagy
- Clinical
- General phenotypes: ALS, ALS + FTD, Juvenile-onset ALS, Madras
- Onset
- Age: 10 to 71 years; Older in females
- Bulbar dysfunction
- Dementia: Some patients
- Upper motor neuron
- Spasticity: Legs > Arms; Gait disorder
- Tendon reflexes: Brisk
- Bulbar & Pseudobulbar dysfunction
- Prominent
- Dysarthria
- Dysphagia
- Lower motor neuron: Often not prominent
- Weakness: Plantar foot flexion; Hands in some patients
- Fasciculations: Some patients
- Wasting: Occasional
- Dementia
- Some patients
- Fronto-Temporal, then Global
- May precede motor signs
- Extrapyramidal: Parkinsonism in some patients
- Sensory: Normal
- Progression
- Slow
- Death after 2 to 17 years
- Laboratory
- CNS pathology
- Motor neurons: Skein-like inclusions
- Cortex & Spinal cord
- Contain Ubiquilin-2, Ubiquitin, p62, TDP43, FUS, Optineurin
- No SOD1
- Spinal cord
- Axon loss: Corticospinal tract
- Anterior horn: Loss of anterior horn cells; Astrocytosis
- Hippocampus
- Ubiquillin-2 pathology in ALS/FTD with & without UBQLN2 mutations
- Aggregates with UBQLN2 mutations often contain: UBQLN2 + p62
- c9orf72 ALS: UBQLN2 aggregated with polyGA DPR protein
- Motor neurons: Skein-like inclusions
- CNS pathology
- Variant phenotype: Madras motor neuron syndrome
- Epidemiology
- ALS 6
36
● Fusion, derived from 12-16 translocation, malignant liposarcoma (FUS; FUS/TLS); Chromosome 16p11.2; Dominant, Recessive or de novo (Sporadic)
- Epidemiology
- Multiple families: More common in Asia
- Familial ALS: Frequency 3% to 5%
- Common in: Early-onset (Juvenile) ALS, Familial or Sporadic
- Sporadic ALS
- Frequency
- General ALS: 0.3% to 0.7%
- Juvenile onset
- 7% to 50%
- More in Japan & China
- Most common gene with fALS onset < 25 years
- Mutations: Exon 6; G507D, R518G, R521C, R521H
- Frequency
- Genetics
- Mutations
- Missense, In-frame deletion or insertion, or Frameshift
- Hot spots: Exon 15; Arg521; Pro525
- Other mutations: > 30; In Exons 3, 5, 6 & 14
- Recessive mutation: H517Q
- R514S & R521C: Associated with Symmetric, Proximal arm, neck & axial weakness
- Pro525Leu & Pro525Arg
144
- Onset age: Very young (<30 years)
- Bulbar presentation
- Disease duration: Short, Rapid progression
- Cytoplasmic inclusions: Stress granule–like
- Pro525Leu: German patients
- Arg521: Less bulbar involvement
- Y526C: Young onset; Rapid progression
- Basophilic inclusions: P525L; c.1554-1557delACAG
- C-terminal nuclear localization truncation mutations: More aggressive disease
- de novo mutations: Described in early onset (11 to 34 years) ALS syndromes
- Incomplete penetrance: Common
- Allelic disorders
- Essential tremor 4
96
- Epidemiology: Rare families
- Mutations: Gln290X; Arg216Cys; Met392Ile; Pro431Leu
- FUS mutant proteins: Degraded by nonsense-mediated-decay pathway
- No symptoms of ALS
- Frontotemporal dementia
- ALS/Parkinson disease
- Juvenile-onset ALS (< 25 years)
- fALS, Recessive
- ALS6, Dominant
- ALS3: ALS family with gene originally localized to 18q21
17
- Essential tremor 4
- Mutations
- FUS protein
177
- Cellular localization
- Normal: Nuclear; Synapses (Pre- & Post-synaptic)
- Disease
- Cytoplasm accumulation
- Present in: Anterior horn cell aggregates in sporadic & hereditary ALS except SOD1
- Protein groups
- Functions
- DNA & RNA metabolism
- DNA-/RNA-binding
- DNA repair
- RNA
- Interacting domains: Located in the mid-region of protein
- Processing: Regulation of splicing & transcription of synaptic mRNAs
- RNA binding: Introns, evolutionary conserved
- Interactions
- Syntaphilin (SNPH)
: Mitochondrial tethering protein
- Mitochondrial proteins
- GluA1 (GRIA1)
- U7 snRNA/snRNP: Regulates expression of histone genes
- snRNP70
- Gene spicing variants
- Alternative 3'UTR selection
- Associated with ALS subtype: Increased oxidative & proteotoxic stress
- Gene spicing variants
- U1 snRNA & U1 snRNP
- Syntaphilin (SNPH)
- Implicated in
- Tumorigenesis
- RNA metabolism
- Stress granule formation
- FUS aggregates: 10% of FTD
- FUS deficient neurons: Decreased spine arborization with abnormal morphology
- Hippocampal neuronal slice cultures: FUS is in RNA granules that are transported to dendritic spines
- Local RNA translation in response to metabotropic glutamate receptor (mGluR5) stimulation
- DNA & RNA metabolism
- Mutations
- Often have abnormal cytoplasm localization
- Disruption of nuclear lamina & nucleoporins 211
- Knockout mice: Perinatal mortality; Male sterility; Chromosomal instability; Radiation sensitivity
- Cellular localization
- Epidemiology
- Clinical
78
- Onset
- Age
- Mean 37 to 44 years; Range Teens to > 70 years
- Variable penetrance at 70 years: 39% to 83%
- Earlier & shorter survival than SOD1 & TDP43 FALS
- Upper cervical & neck
- Limbs or Bulbar: Bulbar more frequent than SOD1 FALS
- Age
- General phenotypes: Predominantly lower motor neuron syndromes
- Axial & arm weakness: Dropped head; Mid to late adult onset
- Benign: Slow progression; Late onset
- Juvenile: Bulbar onset; Cognitive Δ
- Genotype-Phenotype correlations: Mutation locations
- Last 2 amino acids in NLS domain: Aggressive phenotype; Bulbar onset; Cognitive Δ
- Missense at 510 to 521 (K510R, H517Y, Q519E, R521H): Mild; Slow progression; Flail leg
- Missense in C-terminal (Common): Proximal arm; LMN phenotype
- P525L & Y526L: Juvenile
- Lower motor neuron signs
- Common
- Weakness: Distribution
- Limbs
- Bulbar: May be present at onset
- Early: Proximal or bulbar in some patients; Upper or Lower extremities
- May be symmetric or asymmetric
- Posterior neck
- Upper motor neuron signs: Common
- Other CNS
- Cognition: Usually normal; Defecits in few patients
- Extrapyramidal: Parkinsonism in few patients
- Progression
- Variable among families: Range 0.5 to 20 years; Mean 3.4 years
- Slower progression: Later onset age
- More rapid than SOD1 ALS: 89% die in < 4 years
- Neoplasm association: None
- Onset
- Pathology
- Loss of motor neurons: Anterior horn of spinal cord & hypoglossal nucleus
- Myelin pallor in anterior corticospinal tracts
- Macrophages surrounding shrunken Betz cells in motor cortex (neuronophagia)
- Increased lipofuscin staining in neurons
- FUS Variant syndrome: Recessive inheritance
- Epidemiology: Cape Verdean family
- FUS mutation: Homozygous H517Q
- Clinical
- Onset: Proximal upper extremities
- Spread: To lower extremities; Not bulbar region
- Progression: Slow; Up to 14 years
- Variant syndrome: Juvenile ALS with basophilic inclusions
77
- Family history: None
- FUS mutations
- C-terminal region
- P525L (c.1554-1557delACAG); Y526C
- Clinical
- Laboratory
- Pathology
- CNS: Loss of axons in motor roots & corticospinal tracts
- Basophilic inclusions
- In motor & non-motor neurons
- Stain for p62, PABP & FUS
- Tubulofilamentous structures
- Associated with electron-dense granules
- ? Origin from endoplasmic reticulum
- EMG: Diffuse denervation
- Pathology
● Chromosome 20p13; Dominant
- Epidemiology: 1 family
- Genetics
- Linkage security: Probable
- Clinical
- Onset age: Mean = 57 years
- Clinical: ALS = El Escorial criteria
- Course: Progressive; Mean = 3 years
● Vesicle-associated membrane protein B (VAPB)
- Epidemiology
- Southeastern Brazilian families; > 200 patients; Portuguese founder
- Other populations: Rare; Independent European, Asian & American familes described
- Genetics
- Same missense mutation in all families: Pro56Ser
- Likely a mutation hotspot
- Allelic with: Proximal SMA, Adult onset (SMAFK)
- Same missense mutation in all families: Pro56Ser
- VAPB protein
- Location: Associated with ER & Golgi membranes
- Lipid-droplet-ER contact
- Regulates membrane delivery into dendrites.
- Mutant protein: Intracellular aggregates not associated with membranes
- Clinical: 3 variant syndromes
- Onset
- Age: 25 to 55 years
- Cramps
- Fasciculations
- Weakness: Few patients
- Heterogeneity: Inter- & Intra-familial
- Atypical ALS
- Onset: 25 to 44 years
- Bulbar signs: Dysphonia (20%)
- Lower motor neuron
- Weakness
- Leg predominant
- Arms & Legs
- Proximal predominant: 85%
- Trunk & Tongue in some patients
- Cramps: Long-standing; Painful; Easily precipitated
- Fasciculations
- Weakness
- Upper motor neuron features: 30%; No effect on survival
- Tendon reflexes
- Common: Reduced
- Some patients: Present in weak muscles
- Increased: 30%
- Jaw jerk: 20%
- Postural tremor
- Sensory loss: Few patients
- Course
- Slowly progressive
- Survival: Mean 22 years
- Non-invasive ventilation: 7 to 10 years
- ALS, Rapidly progressive
- Survival: < 5 years
- Lower motor neuron signs
- Pyramidal signs
- Occurs in same kindreds as atypical ALS syndrome
- Spinal muscular atrophy, Late onset
- Onset 35 to 55 years
- Weakness: Predominantly proximal
- Fasciculations
- Cramps
- Tendon reflexes: Reduced or Absent
- No bulbar or pyramidal involvement
- Onset
- Laboratory
- Serum CK: Mildly elevated (80%) or Normal
- EMG: Denervation, proximal, distal & tongue; Giant motor unit potentials
- NCV: Normal motor & Sensory
- Muscle biopsy: Neurogenic
- Groups of large & small angulated fibres
- Fiber type grouping
● Angiogenin (ANG)
- Epidemiology
- Most identified in Irish & Scottish populations
- Uncommon in US
- Mutations found in some patients with no family history of ALS
- Genetics
- Missense mutations: Q12L; K17I; K17E; R31K; C39W; K40I; I46V; V113I
- Similar mutations occur in some Parkinson patients
- Some missense mutations found in healthy controls: I46V; K17I
- Angiogenin protein
- Mediator of blood vessel formation
- Promotes endothelial invasiveness needed for blood vessel formation
- Induces vascularization of normal and malignant tissues
- Required for VEGF activity
- Ribonuclease family: Abolishes protein synthesis by hydrolyzing cellular tRNAs
- Induced by hypoxia
- Transported to nucleus after uptake by endothelial cells
- Present in wide spectrum of cells: Especially liver
- Cell localization
- Contains nuclear localization signal
- Secreted protein
- Mutation effects: Loss of function
- Mediator of blood vessel formation
- Clinical
- Onset
- Age range: 27 to 76; Similar to ALS or PD without mutation
- Bulbar onset: 50%
- Spinal onset: 50%
- Pattern: "Typical ALS"; No specific phenotype
- Upper motor neuron
- Lower motor neuron
- Course: 0.8 to 10 year survival
- Parkinson disease
- Often occurs with no ALS
- Some ALS syndrome patients have Parkinson signs
- Onset
● Senataxin (SETX)
- Epidemiology: England; Southern Maryland
- Genetics
44
- Mutations
- Missense
- Locations: T3I, Glu385Lys, Leu389Ser, R2136H
- Differ from stop or truncation mutations in senataxin in AOA2
- Allelic disorders
- Distal HMN with upper motor neuron signs
- AOA2 (Recessive)
- Similar locus to
- Mutations
- Senataxin protein
- Clinical
- Onset age: 2nd decade; Mean = < 6 to 21 years
- Early: Gait disorder
- Weakness
- Distal: Later also proximal
- Hands & Feet
- Usually symmetric
- Bulbar disorders: Infrequent, Dysphagia
- Muscle wasting: Distal
- Upper motor neuron signs
- Hyperreflexia 86%
- Upgoing toes 17%
- Sensory: Normal or Mild changes in some (30%) older patients
- CNS: Ataxia in arms in up to 50%
- Severity: Variable degrees
- Course
- Slowly progressive over decades
- 5th & 6th decades: Wheelchair; Loss of hand function
- Milder phenotype: Mild gait disorder
- Laboratory
- Electrophysiology: Motor neuropathy, Chronic
- EMG: Denervation, Distal > Proximal, Chronic
- NCV
- CMAP amplitude: Reduced
- Distal latencies: Long
- Temporal dispersion: Some patients
- Conduction block: some patients
- NCV: Normal
- Sensory: Normal
- Pathology
- Reduced number of Anterior horn cells, especially lumbar
- Sensory pathways: Posterior column fiber loss; Loss of DRG neurons
- Axon swellings: Roots, Spinal gray, Dentate, Cranial nerves 3 & 4
- Spinal cord atrophy
- Muscle MRI: Fat increased in legs
- Serum CK: Normal or Mildly high
- Colon polyps (25%)
- Knock in mice: CD8 cell clonal increase
- Electrophysiology: Motor neuropathy, Chronic
● C9orf72
- Epidemiology
- Frequency
- High frequency
- Europe & America
- 60% of hereditary ALS-FTD
- 27% to 50% of familial ALS patients
- Finnish ALS patients: 50% of familial; 21% of sporadic
- Sweden
- Jewish
145
- Ashkenazi
- Present in up to 80% of fALS
- More common in bulbar onset sALS
- Have nucleotide repeat expansion on common, unstable risk haplotype
- North African Jews
- Other Jewish fALS Association: OPTN
- Ashkenazi
- C9orf72 expansion in UK population: 1 in 700
- Europe & America
- Low frequency: Germany
- Rare: Native Americans; Iran; Asia; Pacific Islands
- Sporadic (Simplex) c9orf72 ALS patient frequency
- ALS
- General: 0% to 50%
- High in Finland
- China: 0.8%
- FTLD: Sporadic 7% to 10%
- ALS
- FTLD familial: 19%
- Lifetime risk of C9orf72-associated FTLD or ALS: 1 in 2,000
- High frequency
- Sex: Male = Female
- Transmission from females: 1.4x higher
- Frequency
- Genetics
- Mutation: GGGGCC (G4C2) hexanucleotide repeat expansion
- Location
- 1st intron & promoter
- Non-coding region between non-coding exons 1a & 1b
- Structurally diverse
- Hexanucleotide repeat numbers
- Controls
- Range of repeat sizes
153
- Control populations: 2 to 45; Rarely as high as 70
- Unaffected individuals
- General: All < 25
- Size range: 2 to 25
- Most common repeat sizes: 2 or 3
- Other peak lengths: 5 & 8
- ≤ 10: 94%
- Intermediate lengths: 7 to 45
- Finland: Repeat lengths > 20 more common
- Repeat type: Uninterrupted
- Linkage disequilibrium between repeat length & neighboring SNP rs3849942
- Intergenerational repeat change
- Rate: 0.29%
- Intergenerational changes (Sequence instability)
- Usually occur from starting repeat length ≥ 8
- No expansion into disease range
- All occurred on rs3849942A haplotype
- Range of repeat sizes
153
- c9orf72 ALS Patients
- Repeat numbers: Range 250 to 4,400
- Symptoms reported with as few as 20 to 22 repeats
- Somatic instability
- Present
- Different expansion size in varying brain & tissue areas
- No recent shared ancestry
- 70 repeat expansions
- Increased C9orf72 expression
- On typical 200-kb risk haplotype
- May increase to pathogenic size range (~1,750 repeats) across generation
- Controls
- Location
- Associated genetics
- Associated with SNPs: rs2477521; rs3849942 allele A
- Finnish founder risk haplotype: All cases with GGGGCC repeat expansion
- Hypermethylation of CpG island near & 5' of G4C2 repeat
103
- Associated & segregates with G4C2 expansion
- Higher degree of methylation: Shorter disease duration
- No methylation for normal or intermediate alleles (up to 43 repeats)
- Penetrance
142
- Younger ages: Low; Rare < 35 years
- 58 years: 50% penetrance
- Older ages
- 83 years: 75% to 99% penetrance
- Females 2 years older than Males
- Female + Bulbar onset: 3 years older
- ALS: Mildly earlier onset age than FTD
- Spinal onset: 2 years earlier than Bulbar
- Genotype-Phenotype correlations & modifiers
- c9orf72 expansion size
- Greater size in blood
- Correlates with: Older age at clinical onset
- No relation to: Clinical syndrome diagnosis
- Size of repeat in brain: Similar in most c9orf72 mutation patients
- Sporadic ALS
189
- c9orf72 repeat length ≤ 2: Longer survival
- c9orf72 repeat length longer in normal range: Shorter survival with bulbar onset
- Greater size in blood
- Frontotemporal lobar degeneration with TDP-43 pathology (FTLD-TDP)
- c9orf72 mutations are most common cause
- TMEM106B
homozygous minor rs3173615 allele (GG; p.T185S)
108
- Protects (OR 0.26) c9orf72 mutation carriers from developing FTD
- No protection from MND
- Also protective (OR 0.12) of FTLD-TDP
caused by GRN
mutations
- TMEM106B A rs1990622 allele: Later age onset
- ATXN2 intermediary polyQ expansions
116
- Associated with development of clinical signs of both FTD & ALS
- Predispose to: Expression of motor neuron disease within phenotype
- Frequency in c9orf72 carriers: 3%
- c9orf72 expansion size
- c9orf72 expansions can also occur with clinical diagnoses of
- Alzheimer disease (1.2%)
- Sporadic Creutzfeldt-Jakob disease (0.2%)
- Huntington disease-like syndrome (1.7%)
- Nonspecific neurodegenerative disease syndromes (2%)
- Ataxia
- Corticobasal syndromes
- Psychiatric disorders: Suicide may be common 163
- Controls (0.15%)
- r(GGGGCC)n RNA repeats: Possible disease mechanisms:
196
- C9orf72 protein haploinsufficiency (Down regulation)
- Bidirectionally transcribed
- Sense (G4C2) & Antisense (C4G2)
- Into RNA foci: Nuclear
- RNA: Binds to & disrupt RNA splicing, transport & translation
- Non-canonical translation
- Dipeptide repeats (poly-GA, -GP, -GR, -PA, -PR)
- Inclusions or toxicity
- Toxicity
- Directly bind & sequestrate other proteins
- Interfere with: rRNA synthesis, Ribosome biogenesis, translation & nucleocytoplasmic transport
- Mutation: GGGGCC (G4C2) hexanucleotide repeat expansion
- C9orf72 expression
107
- Subcellular: Probably nuclear
- Cellular: Neurons > > Astrocytes or Microglia
- Gray matter > White matter
- Functions through binding to
- Smith-Magenis chromosome regions 8 (SMCR8)
- WD repeat-containing protein (WDR41)
- Regulates autophagy
- G4C2 RNA expression may be downregulated by EXOSC2
- Mutation
- Reduced expression of isoform a
- Associated with formation of nuclear RNA foci
- Clinical
157
- Onset
- Age
- General
- Range: 34 to 84 years
- Mean: 6th decade; 55 to 59 years
- Median: 58 years
- Similar for: ALS & FTD presentations
- Same, or earlier, mean age as: Sporadic ALS
- Young onset (< 45 years): Less than singleton ALS
- Anticipation: 2 to 3 years younger onset per generation 137
- Earlier onset: Sporadic cases 163
- Later onset: More G4C2 repeats
- General
- Clinical
- Weakness
- Limb
- Frequency: 50% to 67%
- Legs: More common
- Arms: Less common
- Bulbar
- Frequency 33% to 43%
- More common than sporadic ALS
- Clustered in some families
- Motor features before Dementia (30%)
- Limb
- Cognitive
- Dementia (FTD) before Motor (32%)
- Psychosis
- Paranoid, deluded or irrational thinking
- Weakness
- Age
- Motor neuron disease
- Occurs in patients without FTD (> 50%)
- Upper motor neuron (95%)
- Tendon reflexes: Brisk (95%)
- Spasticity (46%)
- Plantar response: Upgoing in 20%
- Bulbar dysfunction
- Lower motor neuron
- Weakness
- Fasciculations
- Course
- Survival: Median 30 to 37 months
- Monthly change: -1.4% (sVC) to -1.8% (ALS-FRS)
- vs sporadic ALS: Fewer slowly progressive patients
- No corrrelationi with repeat size
- Clinical variants
109
- No dementia: 50%
- Primarily upper or lower motor neuron involvement: Rare
- Spastic paraparesis: Rare
- Dementia (FTD)
- Frontotemporal lobar degeneration (FTLD)
- Frequency
- Patients: 30% to 35%
- Families: 50%
- Type
- Behavioral (64%)
- Progressive nonfluent aphasia (27%)
- Semantic dementia (9%)
- Psychiatric symptoms: 50%
- Apathetic
- Socially isolated
- Delusion
- Working memory: Impairment
- Less eating dysregulation than FTD non-carriers
- Cortical
- Cognitively impaired
- Limb apraxia
- Cortical sensory loss
- Aphasia: Non-fluent
- May occur in some patients without motor neuron disease
- Extrapyramidal
- Increased tone, Some patients
- Huntington disease phenotype (2%)
- Onset age: Younger than other c9orf72 syndromes; May be pediatric
- Signs: Dystonia, Chorea, Myoclonus, Tremor, Rigidity
- Epilepsy: Myoclonic
146
- Epidemiology: 1 family
- Genetics
- Mutation: c9orf72 repeat expansion
- Other family members with psychiatric or neurodegenerative disorders
- Onset age: 15 years
- Disease duration
- ALS
- Mean 3.6 years; Range 1–6 years
- Long survival: Less frequent than singleton ALS
- FTD: Mean 5 years; Range 1–12 years
- ALS
- Onset
- Laboratory
- EMG: Denervation
- Muscle: Neurogenic atrophy
- MRI
- Atrophy
- Frontotemporal: Bilateral
- Thalamus
- FTD patients: May have presymptomatic cortical thinning
- Fusiform, Thalamic, Supramarginal, Broca & Orbitofrontal regions: Involved in c9orf72, but not sporadic, ALS
- Hypoperfusion: Frontal & Temporal; bilateral
- White matter: Corpus callosum, Superior motor tracts, Corticospinal & Cerebellar pathways
- Atrophy
- CNS pathology
- Brain atrophy: Especially frontal
- Corticospinal tracts: Reduced myelin staining
- LMN: Loss of from brainstem & spinal cord
- Inclusions
- Staining
- Ubiquitin, p62 or TDP-43: Vary in different brain locations
- Ubiquitinated in anterior horn cells, Bunina bodies or Hirano bodies
- TDP-43-positive neuronal cytoplasmic: Cortex & Spinal cord
- Not tau positive
- Locations
- Neocortex
- Hippocampal dentate granule cells
- Cerebellar granule layer
- Intranuclear inclusions
- Transcribed GGGGCC repeat: Forms nuclear RNA foci in cortex & spinal cord
- Proteins: None described
- Staining
● Chromatin-modifying protein (CHMP2B)
- Epidemiology
- Sporadic ALS
- ALS: 1%
- Lower motor neuron predominant ALS (PMA): 10%
- Genetics
- Mutations
- Missense
- Ile29Val; Thr104Asn; Gln206His
- Other (truncation) mutations cause: Fronto-Temporal Dementia
- Mutations
- Protein
- Clinical
- Onset
- Age: 7th & 8th decade
- Bulbar dysfunction
- Lower motor neuron syndrome: (Ile29Val; Thr104Asn; Gln206His)
- Clinical
- Weakness
- Bulbar (Tongue)
- Dysarthria
- Weakness
- Wasting
- Hands or Legs
- Respiratory
- Bulbar (Tongue)
- Fasciculations: Tongue & Limbs
- Tendon reflexes: Reduced or Normal
- No UMN signs
- No FTD
- Progression: Respiratory failure over 15 months
- Weakness
- EMG: Diffuse denervation
- Clinical
- Onset
- Pathology
123
- Motor neurons
- Loss
- Ubiquitylated inclusions
- p62 antibodies (sequestosome 1): Oligodendroglial inclusions in motor cortex
- Neuronal Pathology: Frontal cortex
- Lysosomal: Autofluorescent aggregates
- Motor neurons
- I29V mutation: Fronto-temporal dementia syndrome (FTD-3)
- May be benign variation or pathogenic mutation
- Clinical
- Onset: Dementia
- Spastic dysarthria & dysphagia
- Weakness: 1 arm & both legs
- Tendon reflexes: Brisk
● Oncogene DJ-1 (PARK7; DJ1)
- Epidemiology: Southern Italian & Turkish families
- Genetics
- Mutations: Q45X; E163K & g.168_185dup
- Allelic disorder: Parkinson disease 7, Autosomal recessive, Early-onset
- Clinical
- Onset age: 3rd & 4th decades
- Parkinsonism
- Early onset
- Bradykinesia
- Tremor
- Asymmetric
- Levodopa responsive
- Some families with only PD
- Motor system
- Weakness: Arms & Legs
- Hand atrophy
- Tendon reflexes: Brisk
- Plantar response: Extensor
- Cognitive impairment
- Laboratory
- EMG: Fibrillations; Fasciculations
- NCV: SNAPs normal
- CSF: Normal
● Autosomal Dominant
- Epidemiology: Japanese family
- Clinical
- Onset
- Bulbar dysfunction
- Age: 5th & 6th decade
- Cranial nerves: Atrophy of facial muscles & tongue; Dysarthria
- Weakness: Arms ± Legs; Respiratory
- Tendon reflexes increased
- Normal: Sensation; Intellect; Bladder
- Course: Progressive over 10 years
- Onset
- Pathology
- Reduced numbers of Upper & Lower motor neurons
- Bunina bodies: In lower motor neurons
- Normal: posterior columns
● Neurofilament heavy chain (NEFH)
- Genetics
- Mutation type: Inframe deletion or insertion
- Mutation location
- Large NFH side arm domain
- Hypervariable C-terminus
- Amino acids Lys-Ser-Pro (KSP)
- KSP sequences in normal neurofilament heavy chain
- Repeated > 40 times
- Located in hypervariable region
- Commonly phosphorylated
- Sites for proline-directed serine/threonine protein kinases
- Located in sidearm projection that cross links neurofilaments
- Mouse mutants
- Over expression of neurofilaments: Associated with motor neuron disease
- Neurofilament subunit knock-out: Reduced axon caliber
- Allelic disorder: CMT, Axonal
- Neurofilaments
- Clinical (Genetic associations)
- Sporadic ALS: Mutations in 1% of patients
- Role in disease: ? Risk factor rather than causative effect
- Reduced expression of NFL in sporadic ALS patients without SOD1 or NF mutations
- Tetranucleotide repeat: TTTA in intron
190
- Repeat range: 6 to 15
- 9 TTTA allele: sALS risk reduced; Spinal onset ALS
- 10 TTTA allele; 2.7 yr later onset vs othe sALS
- Hereditary ALS: Scandinavian pedigree
- ALS in 4 members of 2 generations
- Deletion in the NF-H tail: Nucleotides 1989-2030
- Clinical features: Variable
- Dementia
- Dysphagia
- Monomelic ALS
- Sporadic ALS: Mutations in 1% of patients
- NEFH variant syndrome: CMT 2CC, Axonal
129
- Epidemiology: 15 families
- Genetics
- Inheritance: Dominant
- Mutations: Frameshift (Stop) in 3' UTR or C-terminus of NEFH; 1 missense
- Mutation effects
- Translation of cryptic amyloidogenic elements (CAEs) encoded by 3' UTR
- NEFH protein with 40 additional amino acids
- Aggregation-prone NEFH molecule
- Clinical
- Onset age: 2 to 64 years
- Weakness
- Distal or Proximal
- Legs > Arms
- Triceps; Quadriceps
- Muscle wasting: Distal
- Sensory loss
- Varied degrees
- Panmodal
- Distal
- Legs > Arms
- Tendon reflexes: Reduced in legs
- Upper motor neuron: Some patients
- Course: Slow progression
- Laboratory
- Serum CK: Normal to 1,288
- Brain & Spinal cord MRI: Normal
- Muscle pathology
- Denervation: Grouped atrophy; Fiber type grouping
- Internal nuclei
- Rimmed vacuoles: Few fibers
- Mitochondrial oxidative enzymes: Reduced complexes I, II+III & IV
- Nerve pathology
- Axon loss
- NCV: Axon loss, Legs > Arms; CMAP & SNAP amplitudes reduced
- EMG: Chronic denervation, Distal & Proximal; Proximal myopathic changes
- CSF: Normal
- Muscle MRI: Sparing of gracilis & biceps, short head in thigh
● Tar DNA-binding protein (TDP-43; TARDBP)
- Epidemiology
- Families: American, Canadian, English, French, Chinese
- More common in Southern (Sardinia) than Northern European populations
- 33% of ALS in Sardinia (A382T mutation)
- Familial ALS: 3% to 6%
- Sporadic ALS patients: Frequency ~1 in 200; 8 patients described
- 18% with TDP43 mutations sporadic
- Genetics: Mutations
- > 80 identified
- Type: Missense
- Locations
- Often exon 6
- Contains glycine rich domain
- C-terminal
- Interacts with splicing proteins (hnRNPs)
- Most common: A382T
- Other mutations: G287S; Gly290Ala; Ser292Asn; Gly294Ala; Gly298; Ala315Thr;
Glu331Lys; Met337Val; G348C; R361S; G376D; N390D; N390S - G376V: Myopathy
- W385IfsX10 (Stop): Rimmed vacuole myopathy
- C-terminal prion-like domain (PrLD)
- Modulate liquid condensation & aggregation properties of TDP-43
- Other mutation: D169G (Exon 4)
- Clinical Correlations
- Shortest survival: G298S
- Long survival & Late onset: A315T; M337V
- Frequent mutations: G348C; A382T
- Southern China: Gly298Ser
- Often exon 6
- Mutation effects: May increase sTDP-43 isoform
- Allelic disorders
- Frontotemporal lobar degeneration with TDP-43 inclusions, TARDBP-related
- Myopathy with Rimmed vacuoles
- Other
- Frontotemporal lobar degeneration with TDP-43 inclusions, TARDBP-related
- TDP-43 protein
- Expression
- Ubiquitous
- Short isoform (sTDP-43): More prominent in
- Motor neurons
- Cytoplasm
- Subcellular distribution
- Nucleus & Cytoplasm
- Reduced in nucleus in cells with TDP-43 cytoplasmic aggregates
- Properties
- Tendency to self associate & form aggregates
- Sequestered into polyglutamine inclusions
- Phosphorylated TDP-43: Present in neuronal aggregates in ALS & FTD
- Contains: Prion-like domain
- hnRNP protein: Similarity to hnRNPs
- Stress granule related proteins (hnRNP): ALS related
- Tendency to self associate & form aggregates
- Binds DNA & RNA
- Especially in nucleus
- Binds to: Uridine/guanine (UG)-rich motifs in RNA transcripts
- Regulates: Transcription & Splicing
- Lack of RNA binding: May predispose to relocation of TDP-43 to cytoplasm
- Other TDP-43 actions
- Repressor of cryptic exons during splicing
- May be involved in microRNA biogenesis, apoptosis & cell division
- Sporadic ALS: TDP-43 pathology
- Nucleus: TDP-43 reduced
- Cytoplasm: TDP-43 aggregates common
- TDP-43 pathology related to increased mtDNA copy number
- Mutations
- TDP-43 mutation locations: C-terminal in region involved in protein-protein interactions
- Mutation effects
- Loss of TDP-43 from nucleus
- Dysfunctional TDP-43: Inclusion of cryptic exons into mRNAs; RNA stability decreased
- Alternative polyadenylation: Abnormal
- Expression
- Clinical
- General: Onset age earlier; Disease duration longer
- Onset
- Age
- 4th to 8th decade
- Younger than sporadic ALS
- Weakness
- Arms: 50%; More than SOD1 ALS
- Legs: 20%
- Bulbar: Common in Asians
- Age
- Weakness
- Arms before Legs: 60%
- Distal + Proximal
- Respiratory
- Bulbar: Few patients
- Upper motor neuron
- Tendon reflexes: Brisk
- Spasticity: Mild or None
- Cognition
- Usually normal
- FTD-ALS syndromes: Occasional patient
- FTD may be presenting syndrome
- Other family members may have pure ALS with normal cognition
- More involved in Sardinia
- Extrapyramidal: Parkinsonism in few patients
176
- Tremor, resting
- Rigidity
- Akinesia
- Course
- Slowly progressive (5 to 10 years) in most
- Rapid (1 to 2 years) in some families (Gly290Ala; Gly298Ser)
- Mean: Longer survival than sporadic ALS
- Laboratory
- Nerve conduction testing: Normal
- EMG: Denervation, proximal & distal; Fasciculations
- CNS pathology
- Anterior horns of spinal cord: Neuronal loss, Gliosis, and Bunina bodies
- Corticospinal tracts: Pallor
- Betz cell loss in primary motor cortex
- TDP-43 inclusions
- Locations: Upper & lower motor neurons + elsewhere in CNS
- Often ubiquitinated
- Neuronal, Cytoplasmic: Also Nuclear depletion of TDP-43
- Variant: Homozygous A382T missense mutation
80
- Epidemiology: 1 Sardinian patient
- Genetics: A382T missense mutation
- Incomplete penetrance
- Clinical
- Onset age: 49
- Amyotrophic lateral sclerosis
- Bulbar: Dysarthria & Dysphagia
- Spasticity: Arms & Legs
- Tendon reflexes: Increased
- Plantar reflex: Extensor
- Parkinsonism: Rigidity; Bradykinesia
- Dystonia
- Tics: Motor & Vocal; Childhood onset
- Dementia: Frontotemporal
- Laboratory
- EMG: Distal denervation in arms & legs
- NCV: Reduced CMAP amplitude
- Parents: Heterozygous; Normal or Dementia
- TDP-43 variant: Myopathy with Rimmed vacuoles
193
- Epidemiology: 4 families
- Genetics
- Inheritance: Dominant
- Mutations: Frameshift (Trp385IlefsTer10) or Missense (Gly376Val); PrlD/LCD domain
- Protein produced by Frameshift mutation: C-terminal altered PrLD
- Gly376Asp variant: Produces ALS
- Clinical
- Onset age: Adult; 3rd to 7th decade
- Weakness
- Legs > Arms
- Proximal + Distal or Distal predominant
- Forearm: Anterior compartment (Hand & Finger extensors)
- Myalgias
- Exercise intolerance
- Paresthesias: Distal legs
- Course
- Slow progression; Wheelchair 45 to 60 years
- No ALS
- Non-penetrance in 8th decade described
- Laboratory
- Muscle
- Fiber sizes: Varied
- Sarcoplasmic inclusions: TDP-43 & p62 positive
- Endomysial connective tissue: Increased
- Ultrastructure: Autophagosomes
- Transcriptomes: Abnormally spliced sarcomeric genes (TTN & NEB); Increased expression of muscle regeneration genes
- EMG: Motor unit potentials usually small amplitude & polyphasic
- Muscle MRI: Atrophy; Fat replacement
- Serum CK: Mildly high
- Neuropathy: Axon loss in some patients
- Muscle
- Other TDP-43 variant syndromes
- ALS + Extrapyramidal symptoms
- ALS + Supranuclear palsy
- Supranuclear palsy
- FTD without motor neuron disease
- FOSMN
● Peripherin (PRPH)
- Genetics
- Mutations: 4 identified
- Nucleotide insertion in intron 8
- 1-bp deletion in exon 1: Frameshift
- Missense: R133P; D141Y
- Allelic disorder: Axonal neuropathy
- Mutations: 4 identified
- Peripherin protein
- Intermediate filament (type III)
- Location: Peripheral nervous system; Lower levels in motor neurons
- Increased levels after neuronal injury
- Overexpression in mice: Motor neuron disease
- Mutations disrupt neurofilament assembly
- Associated with ubiquitinated inclusions
- Round & Lewy body-like inclusions
- Hyaline conglomerate inclusions
- Axonal spheroids: Occur in proximal axons of diseased motor neurons
- Serum peripherin levels 219
- Clinical: 4 patients
- Onset age: 57 years
- Course: 5 years
- Onset: Asymmetric leg weakness
- ALS diagnosis
- PRPH variant disorder: Sensory-Motor neuropathy
154
- Epidemiology: Icelandic population
- Genetics
- Inheritance: Recessive
- Mutation: Splice-donor variant; c.996+1G>A; Loss-of-function
- Clinical
- Neuropathy (50%)
- Onset age: 20 years
- Sensory
- Pain & Vibration loss
- Subjective: Foot numbness
- Weakness: Toe extensors
- Neuropathy (50%)
- Laboratory
- Sural NCV
- SNAP Amplitude: Smsll
- Conduction velocity: Normal
- Heterozygotes: Partial reduction in sural SNAP amplitude
- Sural NCV
● FIG4
- Epidemiology
- 10 patients of European ancestry
- Frequency: 1% to 2% of ALS patients
- ALS
- Mutations
- Heterozygous
- Missense or Termination
- No patient with Ile41Thr mutation common in CMT 4J
- Allelic disorders
- Mutations
- FIG4 protein
- Clinical patterns
- Onset age: Mean 56 years; Range 29 to 77 years
- ALS or PLS
- Onset site: Bulbar, Legs or Arms
- Brisk tendon reflexes
- Personality change: Mild; 2 patients
- Disease progression
- Slow to Rapid
- Disease duration: Mean 9 years; Range 1 to 29 years
- ALS + FTD: 1 patient
- Laboratory
- EMG: Normal to Moderate denervation
- SSEP: Normal in 1 patient
- Muscle biopsy: Rare atrophic muscle fibers
- Autopsy: Lower motor neuron loss
- Animal model: 'pale tremor' (plt) mouse
● Profilin-1 (PFN1)
- Epidemiology
- < 30 patients
- 3% of FALS
- 0.2% of SALS
- Genetics
- Mutations
- Actin binding region
- A20T; C71G; ?T109M; M114T; M114V; E117G (also in controls); G118V; R136W; Q139L
- 2 singleton patients with no family history
- Knockout: Mitochondrial pathology, Increased mitophagy
- Mutations
- PFN1 protein
210
- Cellular Locations
- Cytoplasmic
- Mitochondrial
- Actin binding: Converts monomeric G-actin to filamentous F-actin
- Formin dependent
- Axonal integrity & Axon transport
- Nucleotide exchange factor
- Binds ATXN2 & VCP
- Mutant proteins
- May form ubiquitinated aggregates: Increased p62 & ubiquitin in neurons
- Alter growth cone morphology
- Altered actin polymerization into filaments
- Altered autophagy & mitochondrial distribution
- Cellular Locations
- Clinical: Similar to SOD1
- Onset age: 4th to 7th decade; Mean > 50 years
- Onset region
- Limbs: All patients, Especially distal legs
- Bulbar: No patients
- Lower motor neuron: Most defecits
- Upper motor neuron: Few deficits; Tendon reflexes in arms may be brisk
- Respiratory: Late in disese course
- Independent walking: Often lost in 1st 2 years
- Cognitive: Normal
- Course: Progressive; Death < 3 years
- Brain pathology
- Motor neurons: Loss; TDP43 & p62 inclusions
- NO PFN1 inclusions
- Corticospinal tracts: Moderate myelinated axon loss
● V-erb-B2 Avian erythroblastic leukemia viral oncogene homolog 4 (ErbB4; HER4)
- Epidemiology: Japanese, Chinese (0.67% of sALS; 1.2% of fALS) & Canadians
- Genetics
- ErbB4 protein
- sALS: ERBB4 immunoreactivity reduced in spinal cord
- Clinical
- Onset
- Age: Adult; 36 to 70 years
- Arm involvement
- Bulbar: 2 patients
- Lower motor neuron
- Weakness: Limbs
- Respiratory insufficiency
- Upper motor neuron
- Cognitive: Normal or ALS + FTD in 1 patient
- Progression
- Slow, usually
- Death: After 1.5 to 20 years
- Onset
● D-amino acid oxidase (DAO)
- Epidemiology: 1 family
- Genetics
- Mutation: R199W
- May be schizophrenia
locus
- DAO protein
- Tissues: Liver, Kidney, Brain (Neurons & Astrocytes)
- Peroxisomes
- Homodimer
- Flavoenzyme
- Oxidizes: D-serine, D-alanine, D-proline
- Mutation effect: ? Dominant negative
- Clinical
- Onset: Hand weakness & wasting
- Upper motor neuron features
- Lower motor neuron involvement
- Bulbar signs
- Disease progression: Rapid; 1 to 2 years
- Age at death: Mean 44 years; Early 40's in 75%; Range 42 to 73 years
- Pathology in obligate carrier
- Motor neuron loss: Cervical & Lumbar spinal cord
- Lateral corticospinal tract pathology
- Frontal & temporal lobes: Preserved
- TDP-43 in motor neurons: Localized to nuclei; No inclusions
- DAO activity in spinal cord: Reduced
● TAF15 RNA Polymerase II, TATA Box-binding protein-associated factor, 68-kD (TAF15)
- Epidemiology: 6 sporadic patients; 0.5% of sporadic ALS
- Genetics
- Mutations
- Type: Missense;
- Location: Exons 13-16; M368T, G391E, R408C, G452E, G473E
- Allelic with: Chondrosarcoma, extraskeletal myxoid
- Mutations
- TAF15 protein
- Contains: Prion-like domain
- RNA recognition motif (RRM)-containing
- Location: Nucleus & Cytoplasm
- TET proteins (translated-in-liposarcoma, Ewing’s & TAF15)
- Mutant protein: Cytoplasmic foci
- Clinical: ALS, Adult onset
- Onset age: 5th to 7th decade
- Weakness: Distal & Proximal; Bulbar; Asymmetric
- Tendon reflexes: Brisk
- Sensation: Normal
- ALS + FTD: 1 patient
- EMG: Denervation
● Ewing sarcoma Breakpoint 1 (EWSR1)
- Epidemiology: 3 sporadic ALS patients
- EWSR1 protein
- Location: Nucleus; Cytoplasm, punctate or granular, in ALS neurons
- Contains: Prion-like domain
- Aggregation prone: Like FUS, TDP-43, TAF15
- Genetics
- Mutations: Missense; G511A, P552L
- Clinical: ALS
- Onset ages: 35 & 50 years
● Tubulin α-4A (TUBA4A)
- Epidemiology: 12 patients
- Genetics
- Mutations: Val7Ile; G34V; T145P; R215C; R320C; R320H; The349Ser; A383T; W407X; Asp438Asn; K430N
- Allelic disorders
- ALS22
- SPAX11: Ataxia ± Spasticity, Dominant or de novo
- CMYO26: Congenital Myopathy with Focal Myofibrillar Disorganization
- Myopathy/Myotubulinopathy
- OZEMA23: Oocyte/Zygote/Embryo Maturation Arrest 23
- CNS: FTD; Parkinson's
- TUBA4A protein
- α-tubulin
- Expression: Ubiquitous; High in brain
- Component of Microtubules
- See
- Mutations
- Destabilize microtubule network
- Reduced repolymerization capability
- Clinical: ALS
- Onset ages: 48 to 71 years
- Spinal-onset
- Upper & Lower motor neuron signs
- Frontal dementia: 3 patients & 1 1st degree relative
- Disease duration: 1 to 7 years
- TUBA4A variant: SPAX11
(Ataxia ± Spasticity)
204
- Epidemiology: 12 patients
- Genetics
- Inheritance: Dominant or de novo
- Mutations: Missense; Pro173Arg in 3 families
- Clinical
- Onset ages: 2 to 60 years
- Cerebellar: Ataxia, Gait; Nystagmus; Dysarthria
- Spasticity (50%): Legs; DTRs brisk; Bladder dysfunction
- Cognitive decline (33%)
- Arm weakness (17%)
- Course: Progressive
- Laboratory
- Brain imaging: Cerebellar atrophy
- EMG: Chronic neuropathic or myopathic
- TUBA4A variant: CMYO26, Congenital Myopathy with Focal Myofibrillar Disorganization
205
- Epidemiology: 2 patients
- Genetics
- Inheritance: Dominant, de novo
- Mutation: Missense; L227F
- Clinical
- Onset: Congenital or Infancy
- Motor delay
- Myopathic face
- Ophthalmoparesis & Ptosis: 1 patient
- Scapular winging
- Tendon reflexes: Reduced
- Course: No progression; Mild improvement
- Laboratory
- Muscle Fibers
- Sizes: Varied
- Internal nuclei
- Focal myofibrillar disorganisation
- Rimmed vacuoles
- Ubiquitin-positive TUBA4A aggregates
- Desmin accumulations
- Ultrastructure
- Membrane-bound vacuoles with many spherical dense particles
- Myofibrillar disruption
- Z-line streaming
- Serum CK: 491 to 644
- MRI: Fat in gluteus & quadriceps
- EMG: Myopathic
- Muscle Fibers
- TUBA4A variant: Myopathy/Myotubulinopathy
228
- Epidemiology: 31 patients; 19 families
- Genetics
- Inheritance: Dominant; de novo; Recessive; Sporadic
- Mutations: Missense
- Mutation locations
- Common exon: 4
- Guanosine triphosphate (GTP)-ase domain: More severe weakness
- C-terminal domains: Milder presentations
- Common mutation locations
- Glu254: Severe phenotype
- Glu284Lys: Mild myopathy; Also described with OZEMA23
- Recessive mutations: Varied phenotypes
- Severe, early onset Myopathy
- Late onset, Multisystem
- Mutation effects: Disrupt microtubule morphology
- Clinical
- Onset: Birth to 60 years
- Weakness: Axial, Proximal, Distal, Face, Ptosis, Respiratory
- Fatigue: Recessive mutations
- CNS: 20%; Ataxia; Epilepsy; Exon 2 mutations
- Laboratory
- Serum CK: 143 to 2,794; Mean 1,500
- Muscle pathology
- Fiber sizes: Varied
- Internal nuclei
- Endomysial fibrosis: 60%
- Autophagic vacuoles: 30%
- Myofibril disorganization: 30%
- Aggregates: p62, TDP-4; TUB4A4
- Rods: 30%
- Spheroid bodies: 30%
- Muscle MRI: Axial muscles most consistently involved
● TANK-Binding Kinase 1 (TBK1)
- Epidemiology
- Frequency: 1% to 3% of ALS & ALS/FTD; > 40 families
- Common in Belgium
- Genetics
- Mutations
- Missense & Stop
- E696K, Glu643del, c.456_457delGT
- Effects: Loss of function; ? Dominant gain of function
- Heterozygous
- Penetrance
- Incomplete
- Family history: Often negative
- Allelic disorders: Different syndromes in same family
- Increased TBK1 copy number: Glaucoma (Primary open angle; Normal tension)
- PLS/Dementia
- Primary progressive aphasia
- Parkinsonism, atypical: Cortico-basal syndrome
- Encephalopathy, acute, infection-induced (herpes-specific), susceptibility to, 8 (IIAE8)
- Autoinflammation + Arthritis & Vasculitis, Recessive (AIARV)
- Mutations
- TBK1 protein
- Kinase family
- Serine/Threonine
- IKK (IκB)
- Autophagy
- Efficient cargo recruitment in autophagy
- Degradation of protein aggregates
- Regulates selective autophagy
- Co-localizes with & phosphorylates OPTN & SQSTM1 in autophagosomes
- NF-κB pathway
- Related to: Innate immunity, type I IFN response
- Inhibits RIPK1
- Kinase family
- Clinical
- ALS syndrome
- Onset
- Age: Mean 60 years; 4th decade to 80 years; Mean 9 years later than c9orf72
- Common: Legs or Arms
- Bulbar in 15% to 40%
- Bulbar involvement: 87%; Dysarthria; dysphagia
- Upper motor neuron: Prominent
- Tendon reflexes: Brisk
- Spasticity: May be asymmetric
- Lower motor neuron
- Fasciculations
- Muscle atrophy
- Onset
- Fronto-Temporal dementia
- Cognitive impairment in 50%
- Language impairment (Aphasia)
- Presenting feature in 60%
- Extrapyramidal features: Hypokinesia; Tremor; Rigidity
- Course
- Survival: Mean 42 months; Range 1 to 9 years
- Age at death: Mean 65 years; Range 50 to 77 years
- ALS syndrome
- Laboratory
- EMG
- Denervation, Active & Chronic
- May be normal
- Myopathic features in some
- Serum CK: Mildly high
- Brain MRI: Atrophy, often temporal & asymmetric
- FDG-PET: Asymmetric hypoperfusion or glucose hypometabolism
- Pathology
- CNS
- Brain atrophy: Frontal & Temporal
- TDP-43 & p62 inclusions: Neurons & Glia
- Motor neuron loss
- Muscle
224
- Denervation: Targets; Angular muscle fibers; Grouped atrophy
- Myopathy in some patients: p62 aggregates; Vacuoles; Endomysial lymphocytes
- CNS
- EMG
- TBK1 variant: PLS/Dementia
140
- Epidemiology: 1 Spanish family, 6 patients
- Genetics
- Inheritance: Dominant
- Mutation: Arg573Gly
- Clinical
- Onset age: 60 to 76 years
- Dementia: 67%
- Behavioral changes
- Aphasia, Non-fluent
- Memory deficits
- Course: Progressive
- PLS syndrome: 33%
- Distribution: Bulbar ± Spinal
- Course: Slow progression
- May occur without dementia
- EMG: No denervation
● Cyclin F (CCNF)
- Epidemiology: 72 patients; Diverse geography; 0.9% of ALS
- Genetics
- Mutations: 43 diffferent; Missense, mostly; S3G, K97R, T181I, S195R, R392T, S509P, T543I, S621G (Segregates), E624K, I772T
- Similar locus to: ALS-FTD 16p12
- CCNF protein
- F-box
- Component of E3 ubiquitin–protein ligase complex (SCFCyclin F)
- Genome stability
- Regulates: Deoxyribonucleotide triphosphate levels, Centrosome duplication, Spindle formation
- Targets & regulates levels of SCFcyclin F substrates
- Effects VCP activity in cytoplasm
- Clinical
- Onset age: Mean 53 years; Range 42 to 66 years
- Syndromes
- ALS alone
- Most common presentation
- Limb or Bulbar (10% to 27%) onset
- UMN + LMN signs
- Asymmetric
- PLS syndrome: 1 patient
- FTD-ALS syndrome: 1 patient
- Disease duration: 3 to 4 years (43 months)
- ALS alone
- Laboratory
- TDP 43 brain pathology: 1 patient
● CYLD Lysine-63 Deubiquitinase (CYLD)
- Epidemiology: European Australian family,
- Genetics
- Mutation: Missense; c.2155A>G, p.M719V
- Allelic disorders
- Brooke-Spiegler syndrome
- Cylindromatosis, familial
- Trichoepithelioma, multiple familial, 1
- Brooke-Spiegler syndrome
- CYLD protein
- Clinical
- Onset age: 30 to 62 years
- ALS
- Weakness: Flaccid paralysis
- Bulbar
- Dysarthria
- Dysphagia
- Jaw jerk: Present
- Tongue: Wasting; Fasciculations
- Sensory: Normal
- Dementia: Memory loss; Behavior problems
- Course: Death 5 to 12 years after onset
- Other occasional: Parkinson disease; Paget
- Pathology
- Fronto-Temporal atrophy
- Tauopathy: Cytoplasmic neuronal reactivity
- phospho-TDP-43 Cytoplasmic neuronal inclusions
- Hippocampal neurofibrillary tangles
- Glial CYLD-immunoreactivity in white matter
|
● Huntingtin (HTT)
|
HTT (PolyQ) aggregates Not in nuclei; Motor cortex 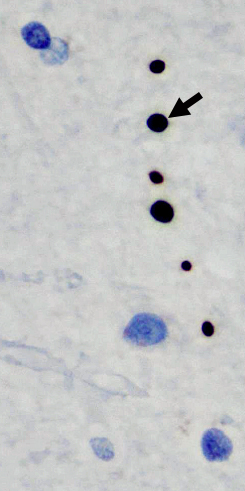 From: R Bucelli; R Hickman (Columbia, NY)
|
● SS18-like gene 1 (SS18L1; CREST)
- Epidemiology: 2 patients
- Genetics
- Possible Mutations: Q388X; I123M; c.660_668del, p.Gln222_Ser224del; c.790G>A, p.Ala264Thr
- SS18L1 protein
- Location: Nucleus
- Calcium regulated transcriptional activator
- Subunit of neuron-specific chromatin remodeling complex (nBAF)
- Neural outgrowth
- Interacts with: FUS
- Clinical
- Onset age: Adult to 8th decade
- ALS syndrome
● Annexin A11 (ANXA11)
- Epidemiology: > 60 patients; 1% to 10% of FALS, 1.7% of SALS
- Genetics
- Mutations
- > 30
- Missense: P36R, G38R, D40G (Common European founder), D40Y, G175R, G189E, R235Q,R346C, G491R
- Splice site
- Penetrance: Incomplete
- Mutations also found in sporadic ALS patients
- Mutations in low complexity domain: More FTD, Bulbar onset, Rapid progression
- Allelic disorders
- ALS23
- Multisystem proteinopathy 6 (MSP6; IBMWMA)
- OPMD syndrome, Child onset
- ALS-FTD
- Corticobasal syndrome (P93S)
- Primary progressive aphasia (D40G) 217
- Sarcoidosis association
- R230C polymorphism associates with Autoimmune disorders & Sarcoidosis
- Sarcoidosis susceptibility (SS3)
- Mutations
- ANXA11 protein
- Calcium binding
- Subcellular location: Nucleus & cytoplasm (vesicle-like structures)
- Binds to: Calcyclin; Phospholipids, RNA granules, Lysosomes
- Roles
- Intracellular trafficking
- Vesicle transport
- RNA
- Granules
- Transport: Long distance with interaction with lysosomes
- Tethering of RNA granules to lysosomes
- Calcium-dependent phospholipid ligand
- Breast cancer & other malignancies: Increased expression
- 56-kD antigen recognized by sera from patients with autoimmune diseases
- Mutated protein
215
- Forms aggregates with: TDP-43 & SQSTM1/p62
- Impaired autophagic flux
- Annexin family
- Calcium-dependent phospholipid-binding proteins
- Bind RNA
- Clinical: ALS
- Onset
- Age: Late; Mean 67 years; Range 37 to 71 years
- Bulbar (80%)
- Weakness
- Bulbar: Dysarthria; Dysphagia
- Limbs
- Flail leg syndrome
- Fasciculations
- Pyramidal signs
- Other CNS: Some patients
- Cognitive impairment (FTD)
- Progressive Supranuclear Palsy
- Course
- Progression: Slow or Rapid
- Age at death: 59 to 85 years; Later with D40G mutation
- Onset
- Laboratory
- Brain pathology
- Neurons: Inclusions & Loss
- Cytoplasmic ANXA11, Phospho-TDP-43 & p62
- Morphology: Skein-like, Tubular-shaped, Filamentous & Complex basket-like
- Brain locations: Motor cortex; Occipital lobe
- Corticospinal tract: Axon loss
- Astrocytes: Increased calcyclin
- Neurons: Inclusions & Loss
- EMG: Denervation
- Brain MRI: Cortical atrophy
- Brain pathology
- ANXA11 variant syndrome: Multisystem proteinopathy 6
174
- Nosology: Inclusion body myopathy + Brain white matter abnormalities (IBMWMA)
- Epidemiology: Brazilian & Greek families, 18 patients
- Genetics
- Inheritance: Dominant
- Mutations: D40Y (Also found in ALS patient); D40V; D40I
- Clinical
- Onset age: Usually 5th or 6th decade
- Weakness
- Asymmetric
- Legs > Arms
- Proximal ± Distal
- Axial: Head ptosis; Abdominal; Scapular winging
- Gastrocnemius: Medial
- Face: Ptosis, mild
- Bulbar
- Gait disorder
- Course: Slow progression
- ALS: 2 patients
- Spinal onset
- Course: Rapid progression
- Fronto-Temporal Dementia (20%)
- No Paget disease of bone
- Laboratory
- Muscle pathology
- Rimmed vacuoles
- Aggregates: Annexin A11, TDP-43, p62, Myotilin
- type 2 fiber atrophy
- No inflammation
- Ultrastructure: Myofibrillar Δ Autophagic material
- EMG
- Myopathic ± Chronic denervation: May be presymptomatic
- Fasciculations: ALS syndrome patients
- Muscle MRI
- Arms: Anterior (Biceps) involvement
- Thigh: Adductors; Posterior (Semitendinosus, Semimembranosus)
- Leg: Gastrocnemius, medial
- Paraspinous
- Brain imaging
- White matter damage: Corticospinal tracts; Frontal subcortical
- Muscle pathology
- ANXA11 variant syndrome: Oculopharyngeal syndrome, Childhood onset
223
- Epidemiology: 1 patient
- Genetics
- Inheritance: de novo
- Mutation: Missense; Asp40Ile; Heterozygous
- ANXA protein
- Mutation effect: Aggregation propensity increased
- Clinical
- Onset age: 1st decade
- Opthalmoparesis
- Bulbar: Dysphonia; Dysphagia
- Weakness
- Neck flexor: 1st decade
- Face
- Axial
- Respiratory
- Limbs
- Arms & Legs
- Proximal & Distal
- Most severe: Foot dorsiflexion
- Scapular winging
- Course: Progressive
- Laboratory
- Muscle pathology: Myopathy; ANXA11 & Osmophilic aggregates; Rimmed vacuoles
- MRI: Fat replacement of muscle; Most in legs
- Serum CK: Normal then 950
● Glycosyltransferase 8 domain-containing protein 1 (GLT8D1)
- Epidemiology: 14 patients
- Genetics
- Mutations: Exon 4; R92C (Common; Short survival), I70T (Long survival), G78W (Long survival), A82E, I87N
- Additional ARPP21
mutation (P529L): Shorter survival
- GLT8D1 protein
- Glycosyltransferase
- Widely expressed: Neurons; Golgi localization signal
- Mutated proteins: Reduced glycosyltransferase activity
- Clinical
- Onset ages: 33 to 66 years; Average 49 years
- Weakness: Limb or Bulbar
- Disease duration: 6 to 101 months
● cAMP-Regulated phosphoprotein 21 (ARPP21)
- Epidemiology
- 7 families; 10 patients
- La Rioja province in Spain
- Genetics
- Mutation: Pro529Leu; Heterozygous
- Mutations associated with risk haplotype
- ARPP21 protein
- Cell location: cytoplasm; Perinuclear & stress granules
- RNA-binding
- Widely expressed in brain
- Regulates: Calmodulin-dependent kinase-1 (CAMK1; 604998)
; Protein phosphatase-2B (PP2B)
- Clinical
- Onset
- Age: 59.5 years
- Region: Spinal > Bulbar
- Pyramidal signs
- Lower motor neuron: Arm & Leg weakness
- Cognitive Δ: 1 of 10 patients
- Course: Mean survival 16 months
- Onset
- Heterogeneous category of: Non-SOD linked; Adult onset; Hereditary ALS syndromes
- Comprises 30% to 50% of dominant ALS syndromes
ALS SYNDROMES: RECESSIVE
- ALS 2: Childhood-onset ALS: Long survival; Usually Recessive
● Alsin (ALS2); Chromosome 2q33.1; Recessive
- Genetics
16
- Mutations
- Disease related: Carboxyl-terminal
- ALS2: Deletions in exon 3 or 4
- Disrupt reading frame
- Allelic with
- Juvenile PLS: Caused by different mutation in same gene
- SPG, Infantile onset
- Juvenile ALS + Dystonia
- Mutations
- Protein
40
- Contains 3 guanine-nucleotide exchange factor (GEF) domains for GTPases
- Localization
- Cytoplasmic face of endosomal membranes
- Requires amino-terminal "RCC1 (regulator of chromatin condensation)-like" GEF domain
- Possible functions
- ? Involved in membrane-proximity activities of small GTPases
- ? Related to vesicle transport & intracellular trafficking
- Tissue Expression
- Neurons in brain & spinal cord
- Low abundance in all tissues
- Mutants
- All have short half-life
- Rapidly degraded by proteasomes
- Clinical features (Type 3)
- Onset age: Mean 6.5 years; Most often < 10 years; Occasional 3rd decade
- Spasticity: Face; Limbs (Paraplegia)
- Pseudobulbar affect
- Amyotrophy: Mild; Distal; Legs > Arms; Increases with age
- Sensory & Bladder: Normal
- Progression: Very slow; Reduced Walking after age 40
- Laboratory
- CSF protein: Normal
- Nerve conduction velocities: Normal
- Nerve biopsy: Mild Reduction in number of large & small axons
- ALS2 variant syndrome: Juvenile ALS + Dystonia
110
- Epidemiology: 8 patients, 3 families
- Genetics
- Inheritance: Recessive
- Mutations: Nonsense; Fameshift; Splice
- Clinical
- Onset age: 1 to 3 years
- UMN
- Spastic diplegia
- Dysarthria
- Gait disorder
- Tendon reflexes: Brisk; ankle clonus
- Cortical
- Developmental delay
- Emotional lability
- Weakness: Generalized
- Muscle wasting: Legs > Arms; Distal > Proximal
- Dystonia: Generalized
- Skeletal: Microcephaly; Scoliosis; Limb contractures
- Other
- Ataxia
- Anarthria
- Course: Bedbound at 5 to 19 years
- Normal: Hearing; Eye movements
- Laboratory
- Muscle pathology
- Small angular muscle fibers
- Fiber type grouping
- NCV: Normal
- Muscle pathology
- Genetics
16
- ALS 5: Childhood-onset
● Spatacsin;
Chromosome 15q21.1; Recessive
- Epidemiology
- 10 families
- ? Most common form of recessive ALS
- Genetics
70
- Spatacsin mutations
- Multiple
- Missense common; Some frameshift
- Many other families linked to similar locus
- Consanguinous families
- Allelic disorders
- Spatacsin mutations
- Spatacsin protein
- Clinical: Weakness, Arm atrophy & Spasticity in all limbs (Type 1)
- Onset age: Range 7 to 23 years; Mean 16
- Spasticity
- Limbs
- Face & Jaw
- Bulbar
- Hyperreflexia
- Plantar responses: Extensor
- Gait
- Amyotrophy & Weakness: Occasionally predominant
- Distal
- Hands & Feet
- Tongue
- Fasciculations
- Course
- Slowly progressive
- Survival: Duration 27 to 40 years
- Cognition: Normal
- Bladder: Normal
- Laboratory
- EMG: Denervation
- Positive sharp waves
- Fibrillations
- Fasciculations
- Nerve conduction
- CMAP amplitudes: Small
- SNAP amplitudes: Normal
- NCV: Normal
- Pathology
- Pyramidal tracts: Small
- Anterior horn cell loss
- Sural nerve: Reduced number of Myelinated axons
- Corpus callosum: Normal
- CSF protein: Normal
- EMG: Denervation
- Epidemiology
- ALS 12: Adult-onset
74
● Optineurin (OPTN);
Chromosome 10p13; Dominant or Recessive
- Epidemiology
- Japanese consanguinous, German, Italian & Dutch families
- 38 patients described
- 1% to 4% of FALS
- SALS: < 1%
- Genetics
- Mutations
- Japanese: Recessive or Dominant; Exon 5 homozygous deletion, Q398X homozygous, E478G heterozygous
- Other: All Dominant; Missense, Nonsense, Intronic
- Common location: C-terminal Ubiquitin-binding domain
- Possible mutation mechanism: Haploinsufficiency
- Allelic with: Primary open-angle glaucoma
- Most mutations: Single copy missense
- Possible mutation mechanism
- Overactivate OPTN-TBK1 pathway
- Cause increased OPTN & TBK1 phosphorylation & activity
- Mutation with either ALS or glaucoma
- 691_692insAG: Dominant or Recessive ALS
- Mutations
- OPTN protein
- Interacts with: Huntingtin
;
Transcription factor IIIA;
RAB8
- Colocalises with SOD1- and TDP-43-positive inclusions in ALS
- Binds to ubiquitinated receptor-interacting protein
- Negatively regulates TNF-α induced activation of NF-κB
- Autophagy-related
- Interacts with: Huntingtin
- Clinical
- Onset
- Age: Range = 24 to 83 years; Mean = 62 years
- Weakness: Legs > Bulbar, Arms
- Weakness
- Arms
- Legs
- Asymmetric
- Respiratory: Late in course
- Bulbar
- Some patients
- Dysphagia; Dysarthria; Tongue fasciculations
- Tendon reflexes: Normal or Increased
- Spasticity: Prominent in 70%
- Progression: Variable
- Range: 0.75 to 25 years
- Mean: 5 years
- Rapid progression: Q165X; Q454E; K557T; p.Lys440Asnfs*8
- Fronto-Temporal Dementia: Few patients
- No glaucoma
- Onset
- Laboratory
- EMG: Diffuse denervation
- Pathology
- Spinal cord: Loss of myelin & Axons
- OPTN cytoplasmic inclusions
- Epidemiology
- ALS 16: Childhood-onset
81
● Sigma-1 Receptor (SIGMAR1; σ1R);
Chromosome 9p13.3; Recessive
- Epidemiology: Eastern Saudi Arabian family
- Genetics
- Mutation: Homozygous; Missense; E102Q
- Allelic disorders
198
- ALS, Adult onset, Sporadic
- Parkinsonism-ALS, Adult onset,
- Distal HMN (dHMN)
- ? FTD-MND
- Distal SMA2 (Jerash type, DSMA 2; HMNJ): Upper motor neuron signs common
- SIGMAR-1 protein
- Type II membrane protein: Short cytosolic N-terminal tail 180
- Cell location: Endoplasmic reticulum (ER)
- Expression areas: Ubiquitous, High in Motor neurons
- Sub-cellular: Post-synaptic cholinergic densities (C-terminals)
- Chaperone
- Ligand: Neurosteroids, Psychostimulants, Dextrobenzomorphans, Dimethyltryptamine
- Functions
- Ion channel modulation
- Interacts with K+ channels & Inositol 1,3,5-triphosphate receptors (IP3Rs)
- Lipid transport
- Neuronal cell differentiation
- ER stress response
- Regulates: Axon elongation; Tau phosphorylation
- Ion channel modulation
- Mutation effect: Abnormal subcellular distribution of SIGMAR-1
- Clinical
- Onset
- Age: 1 to 30 years
- Spasticity: Legs
- Spasticity
- Legs > Arms
- Tendon reflexes: Brisk
- Weakness
- Hand & Forearm muscles: Especially extensors & triceps
- Normal: Sensation; Cognition; Bladder
- Progression: Often to wheelchair by 3rd decade
- Onset
- Laboratory
- EMG: Denervation in limb muscles; Motor units large
- NCV: Normal velocities
- Brain MRI: Normal
- SIGMAR1 Variant syndrome, Possible: Frontotemporal lobar degeneration-Motor neuron disease
- Inheritance: Dominant
- Epidemiology: Australian families
- Genetics
- 3' UTR point mutations: Some may be polymorphisms, also occur in normals
- Some patients & familes: Pathogenic mutation may be another gene, c9orf72 expansion
- SIGMAR1 Variant syndrome: Distal hereditary motor neuropathy (HMNR2; dHMN; DSMA2)
122
- Epidemiology: Chinese (3 patients), Portugal & Afghan families
- Genetics
- Inheritance: Recessive
- Mutations: Deletion, or Splice
- c.151+1G>T: Produces shortened protein (σ1R31_51del); Homozygous
- c.561_576del (Exon 4; Stop)
- Exon 4 deletion
- Similar locus to: Distal SMA2 (Jerash type, DSMA 2; HMNJ)
- Clinical
- Onset age: Range 5 to 12 years
- Weakness
- Distal
- Legs > Arms: Foot drop
- Symmetric
- Wasting: Distal; Legs > Arms
- Tendon reflexes
- Ankles: Absent
- Knees: Increased or Reduced
- Plantar reflex: Extensor
- Feet: Pes equinovarus; Hammer toes
- Sensory: Normal
- Course: Slow progression, more rapid during adolescence
- Laboratory
- NCV
- Velocities: Borderline (32 to 52 M/s); May vary among nerves
- CMAPs: Amplitudes reduced; Temporal dispersion in some
- SNAPs: Normal
- EMG: Distal denervation; Large motor units
- Sural nerve: Normal
- Brain MRI: Normal
- NCV
- ALS
188
● Ring finger Protein 13 (RNF13);
Chromosome 3q25.1; Recessive
- Epidemiology: 1 family; 3 patients
- Genetics
- Mutation: Homozygous; Intron; c.195+1G>A
- Allelic disorder: Developmental and epileptic encephalopathy 73 (DEE73)
- RNF13 protein
- Mediator of endoplasmic reticulum stress response & apoptosis
- Interactions: ERN1
; JNK
- Neurite outgrowth: Stimulation
- Mutation effect: Abnormal RNF13 splicing
- Clinical
- Onset ages: 4th & 5th decade
- Weakness: Arms, then legs
- Upper motor neuron: DTRs increased; Leg spasticity in 2 patients
- Extrapyramidal: Bradykinesia; Tremor
- Sensation: Normal
- Course: Slow progression
- Laboratory
- EMG: Denervation, diffuse
- Brain MRI: White matter - Periventricular hyperintensities, Corpus callosum atrophy
- ALS: Childhood-onset
64
● Chromosome 6p25, 21q22; Recessive- Epidemiology: 1 Utah family, 4 patients
- Clinical
- Onset age: 4 to 10 years
- Ptosis: Mild
- Weakness & Atrophy
- Distal
- Arms & Legs
- Symmetric
- Respiratory failure: Late in course, 1 patient
- Bulbar & Cranial
- Speech: Dysarthric; Spastic
- Dysphagia
- Face weakness, mild
- Upper motor neuron
- Spasticity: Legs > Arms
- Tendon reflexes: Brisk in legs ± arms
- Plantar response: Upgoing or absent
- Sensory loss: Mild; Distal; Vibration
- Gynecomastia: 1 patient
- Progression: Slow; Loss of ambulation after decade
- Laboratory
- NCV
- Normal velocities
- CMAPs: Reduced amplitude in legs
- SNAPS: Reduced amplitude
- EMG: Denervation, Distal
- Nerve biopsy: Minimal loss of sensory axons
- Muscle biopsy: Grouped atrophy; Fiber type grouping
- NCV
- Recessive ALS: Other types
- Clinical syndrome: Weakness & severe spasticity, esp. in legs (Type 2)
- Symptoms in lower limbs
- ? form of familial spastic paraplegia
- Clinical syndrome: Weakness & severe spasticity, esp. in legs (Type 2)
BULBAR MOTOR NEURON SYNDROMES: Hereditary & Other
|
Brown-Vialetto-van Laere 1: SLC52A3; 20p13; Recessive 2: SLC52A2; 8q24; Recessive 3: UBQLN1; 9q21; Dominant Bulbar ALS Fazio-Londe: SLC52A3; 20p13; Recessive Kennedy's Syndrome: Androgen Receptor ; Xq12; Recessive Madras motor neuron disease: Sporadic Spino-bulbar muscular atrophy: Dominant Worster-Drought |
Fazio-Londe
● SLC52A3 (RFT2; c20orf54)
- Definition
- Progressive bulbar palsy of childhood
- Disorder of bulbar motor neurons
- Genetics
- Allelic with: Brown-Vialetto-van Laere 1 (BVVLS1)
- Clinical features
- Onset: 1 to 12 years
- Cranial nerves
- Ptosis
- Facial weakness
- Dysphagia
- Normal hearing
- Respiratory stridor
- Hyperreflexia
- Subtypes
- Dominant: Rare
- Recessive: Early onset
- Respiratory failure
- Rapid progression: Death < 2 years from onset
- Recessive: Later onset
- Less respiratory involvement
- Bulbar: Dysarthria; Dysphagia
- Facial weakness
- Slow progression
- Laboratory
- CSF: Normal
- Serum CK: Normal
- EMG: Denervation; Large amplitude Motor unit potentials
- Neuropathology
- Loss of neurons from motor cranial nerve nuclei
- Occasional: Anterior horn cell loss; Cerebelar involvement
- Similar to: Brown-Vialetto-van Laere
- But no hearing loss
Brown-Vialetto-van Laere 1 (BVVLS1)
● SLC52A3 (RFT2; c20orf54)
Brown-Vialetto-van Laere 2 (BVVLS2) ● SLC52A2 (RFT3; GPR172A; RFVT2)
Brown-Vialetto-van Laere 3 (BVVLS3) 94 ● Ubiquilin 1 (UBQLN1)
|
|
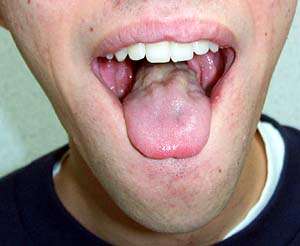
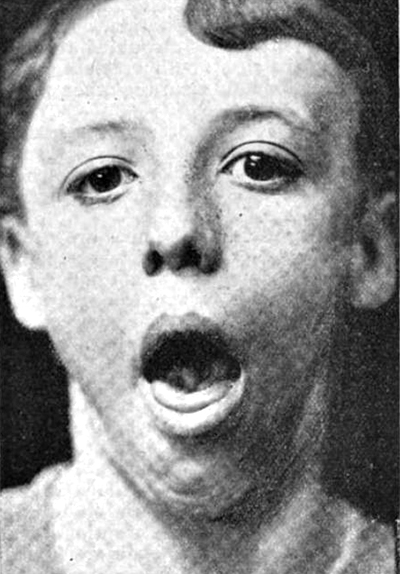
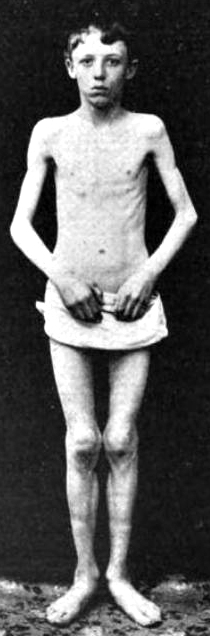
Madras Motor Neuron Disease 61
● Sporadic; Occasional recessive inheritance
- Epidemiology
- Common in South India
- Family history of similar disorder: 2%
- Male = Female
- Genetics
- A8302G mutation in mt-tRNALys
100
- Homoplasmic in 7% of Madras patients
- High levels (82%) also in some unaffected family members
- UBQLN2: M392L Mutation
- No mutations in: SLC52A1, SLC52A2, SLC52A3, c9orf72
- A8302G mutation in mt-tRNALys
100
- Clinical
- Onset
- Age: Juvenile; Mean 16 years, Range 1st to 5th decade
- Hearing loss: 53%
- Arm weakness: 38%
- Cranial nerve
- VII: Facial diplegia, Lower > Upper
- VIII: Deafness, Sensorineural (30% to 80%)
- IX to XII: Bulbar involvement (40%)
- Dysphagia
- Dysarthria (Hoarseness)
- Tongue: Weakness; Fasciculations
- Lower motor neuron signs (85%)
- Weakness
- Limbs: Arms > Legs
- Asymmetric
- Diffuse: Distal > Proximal
- Wasting
- Fasciculations: Limbs & Face
- Weakness
- Pyramidal signs (80%)
- Reflexes
- Tendon reflexes: May be brisk in legs
- Plantar: May be extensor (50%)
- Sensory: Normal
- Other: ? Minipolymyoclonus related to fasciculations
- Course
- Slowly progressive
- Most remain ambulatory
- Onset
- Laboratory
- Electrodiagnostic
- EMG: Active & Chronic denervation in cranial, proximal & distal muscles
- CMAPs: Small amplitude
- SNAPs: Normal
- Pathology: 1 patient
- Anterior horn cells: Reduced
- Ventrolateral column: Axonal loss
- Other cranial nerve nuclei involved: 3, 5, 6, 7, 10
- Cochlear nuclei: Neuronal loss
- Other tracts involved: Spinocerebellar & Spinothalamic
- Other regions of CNS also involved
- Cellular: Microglial reaction; No cytoskeletal pathology
- Electrodiagnostic
- Differential diagnosis: Similar to
- Variant: Madras MND + CNS features (13%)
33
- Epidemiology: India
- Family history
- Positive: 29%, More frequent than typical MMND
- Clinical
- Onset age: Younger than typical Madras MND
- Bulbar dysfunction (100%): More common than typical Madras MND
- Optic atrophy (100%)
- Cerebellar disorder: Ataxic gait
- Pyramidal signs
- Minipolymyoclonus
Worster-Drought syndrome: Congenital suprabulbar paralysis
- Inheritance patterns
- X-linked
- Autosomal dominant with reduced penetrance
- Clinical
- Course
- Onset: Congenital
- Age at diagnosis: 6 years
- Non-progressive
- Male > Female: 3:1
- Familial cses: 6% to 21%
- Bulbar paralysis
- Abnormal tongue & lip movement (98%)
- Brisk jaw jerk (90%)
- Airway obstruction: Especially neonatal presentation
- GI: Dribbling; Poor nutrition; Aspiration; G-E reflux
- Speech: Dysarthric
- Spastic tetraparesis (91%): Mild
- Cortical
- Language disorder: Expressive > Receptive
- Learning difficulties (81%)
- Neuropsychiatric disorders (41%)
- Epilepsy (28%): Neonatal; Febrile; epilepsy
- Congenital defects (61%)
- Contractures: Jaw & Other
- Palatal
- Micrognathia
- Fused teeth
- Eye movements: Abnormal (13%)
- Course
- Laboratory
- CNS Imaging
- Abnormal in 32%
- Perisylvian polymicrogyria: Unilateral or Bilateral
- Swallowing studies: Abnormal, especially in oral & pharyngeal phases
- CNS Imaging
Cortical Suprabulbar palsy
- Foix-Chavany-Marie syndrome: Faciopharyngoglossomasticatory diplegia
- Loss of voluntary control: Preserved automatic function
- Anatomy: Bilateral persylvian lesions
Multisystem disorders
- Hexosaminidase A: GM2 Gangliosidosis, Late Onset
● β-Hexosaminidase A (HEXA)
;
Chromosome 15q23; Recessive
- Nosology: Tay-Sachs, Late onset (LOTS)
- Genetic features
- Gly269Ser substitution in α chain
- Most common mutation in adult form
- Usually in compound heterozygosity with null mutation
- Most common mutation in adult form
- Other mutations with milder disease
- Missense: Arg499His; Arg499Cys; Arg504His; Arg504Cys
- Splice site: IVS7-7G
- General: Chilhood + Adult mutations
- Other mutations (null): More severe disease
- Exon 11: 4-bp insertion
- Exon 12: G to C mutation in splice junction
- Exon 1: A to T in initiation codon
- Exon 11: 4-bp insertion
- Misdiagnosis
- Pseudo-deficiency alleles: Arg247Trp; Arg249Trp
- False negative: B1 variant; Abnormal reactivity to GM2 but not artificial substrate
- Gly269Ser substitution in α chain
- HEXA protein
- Location: Lysosome enzyme
- Function
- Breakdown of gangliosides
- Hydrolyzes GM2 ganglioside to GM3
- Disease syndromes: < 10% of normal Hex A activity
- Later-onset syndromes: Higher Hex A activity
- Clinical features
- Onset: Cramping in legs
- Weakness
- Proximal > Distal
- Selective: Triceps; Quadriceps
- Symmetric
- May be only sign, or present with CNS features
- Spontaneous activity
- Cramps
- Fasciculations
- Cerebellar
- Gait Ataxia
- Dysarthria
- Nystagmus
- Tremor
- Cortical function
- Dementia
- Manic-Depressive-like disorder
- Easy startle
- More common with neuropsychometric testing: Subcortical
- Deficits: Attention, Processing speed, Executive function, Memory
- May be normal
- Sensory: Unusual
14
- Paresthesias: Distal; 2nd decade onset
- Loss: Mild; Pansensory; Distal legs
- Tendon reflexes: Normal, Reduced in legs, or Brisk
- Corticospinal: Some patients
- Spasticity
- Bladder dysfunction
- Movement disorder: Dystonia & Choreoathetosis
- Cranial nerves: Spared
- Exacerbating drugs: Psychotropic medications (Haloperidol, Chlorpromazine, Risperidone)
- Lab
- MRI: Cerebellar atrophy common
- Nerve biopsy: Axon loss, Sensory & Motor
- Muscle biopsy: Denervation, Grouped atrophy, Pyknotic nuclear clumps
- NCV: Small amplitude CMAPs & SNAPs
- EMG: Large motor units; Fibrillations; CRDs
- Autonomic pathology: Membranous cytoplasmic bodies (MCBs) in neurons
- Experimental treatments
- Lower motor neuron phenotype also with Sandhoff disease
- Machado-Joseph
(esp Type I)
● CAG triplet repeats; Chromosome 14q32.12; Dominant- Clinical features
- Type I = Earlier onset (20 to 30 yrs) with longer CAG repeats
- Dystonia; Facial & Lingual Fasciculations; Bulging Eyes
- Cerebellar signs with distal amyotrophy in later onset (Types II & III)
- Allelic with Spino-Cerebellar Ataxia (SCA) III
- Type I = Earlier onset (20 to 30 yrs) with longer CAG repeats
- Clinical features
- Polyglucosan body disease
: Adult-form
● Glycogen branching enzyme (GBE1);
Chromosome 3p12.2; Recessive
- Epidemiology
- Frequency: 1 in 600,000 to 800,000
- Ashkenazi Jewish patients: Increased fequency; Carrier 1 in 48
- Non Ashkenazi Jewish: German, Italian, Latin American, Pacific Islander, Caucasian & Cambodian
- Genetics
- Mutations: Missense
- Ashkenazi Jewish patients: Tyr329Ser
- German & Italian: Arg515His & Arg524Gln
- Allelic disorders: GSD 4
- Mutations: Missense
- GBE1 protein
- Clinical features: CNS + Polyneuropathy
39
- Onset
- Age
- Adult: 20 to 71 years; Mean 51 years
- Other forms in childhood: Myopathy & Hepatic disease
- Neurogenic bladder
- Age
- Polyneuropathy
- Legs > Arms
- Motor: Weakness, Distal
- Sensory loss: Large fiber modalities (Vibration)
- Autonomic: Impotence; Orthostatic hypotension; Fecal incontinence
- Muscle
- Weakness (50%): Proximal
- Muscle MRI: Glutei; Semimembranosus; Biceps femoris; Gastrocnemius
- Myelopathy
- Spasticity
- Legs > Arms
- Gait disorder
- Tendon reflexes: Brisk in legs
- Plantar responses: Extensor
- Neurogenic bladder
- Spasticity
- Dementia & Cognitive (50% to 67%)
- Cortical & Subcortical
- Attention & Memory defecits
- Course
- Bladder Δ → Gait disorder → Cognitive decline
- Survival: Mildly reduced; Mean 70 years
- Atypical features in some patients
- Earlier onset
- Ataxia
- Parkinsonism
- Cardiomyopathy, dilated
- Course
- Progression: Slow
- Onset
- Laboratory
- MRI
- Cortical atrophy
- White-matter changes (Leukodystrophy): Periventricular; Subcortical
- Distribution: Bilateral; Symmetric; Occipital
- Cavitary changes: Internal capsule; Anterior perforated substance
- FLAIR: Hyperintense pial rim; T2 hyperintense mesencephalic ‘beak’
- Corpus callosum: Thin
- Diffuse atrophy: Brain & Spinal cord
- Nerve conduction studies: Neuropathy
- Axon loss
- Sensory & Motor involvement
- Serum CK: May be high
- Liver enzymes (AST & ALT): May be mildly high
- GBE activity: Reduced by 60% to 80%
- Muscle fiber pathology
- Basophilic aggregates
- PAS
- Dense positive, α amylase resistant material
- Polyglucosan accumulation
- MRI
- Nerve & CNS Pathology
- Dx by sural nerve or axillary skin biopsy
- Polyglucosan bodies
- Other locations: Apocrine glands of axillary skin; Skeletal muscle; Peripheral (sural) nerve
- Perivascular inflammation: Occasional
- Epidemiology
- Also see: ALS with polyglucosan bodies
● ALDH18A1 (P5CS)
- See SPG 9A (ALDH18A1)
- Clinical features
- Onset: 1 to 37 years
- Cataracts
- Weakness
- Distal arms & proximal legs
- Exacerbation during pregnancy; remission afterwards
- Distal arms & proximal legs
- Upper motor neuron signs
- Skeletal
- Short stature; Dysplastic skull base; Small carpal bones
● Microtubule-associated protein tau (MAPT)
- Allelic with
- Frontotemporal dementia with parkinsonism
- Frontotemporal dementia with motor neuron disease
- Progressive supranuclear palsy
- Tauopathy + Respiratory failure (Recessive)
- Pick disease
- Lower motor neuron disorder
- Parkinson-ALS
- Parkinson susceptibility
- Clinical
- Onset: Mean age 45 years; Alcoholism or Depression
- Dementia: Behavior & judgement > Language & praxis; Kluever-Bucy syndrome
- Disinhibition: Alcoholism; Hyperreligiosity; Excessive eating, sexual behavior
- Parkinsonism
- Amyotrophy
- Variability of symptoms among patients
- Pathology
- Superficial cortex: Spongiform changes
- Spinal cord: Motor neuron loss
- MAPT variant syndrome: Lower motor neuron syndrome
112
- Epidemiology: 1 family, 5 patients
- Genetics
- MAPT mutation: D348G
- Inheritance: Dominant
- Clinical
- Onset age: 6th & 7th decade
- Weakness
- Proximal: Arms
- Respiratory
- Tendon reflexes: Reduced
- Leg cramps
- Fatigue
- No: Frontotemporal lobar degeneration; Semantic dementia; UMN features
- Pathology
- α-motoneurons
- Loss
- Phosphorylated tau: Accumulation in surviving motor neurons
- Spinal anterior horns: Atrophy
- α-motoneurons
- MAPT variant syndrome: FTD + Motor neuron syndrome
- Genetics
- Mutation: L317M
- Inheritance: Dominant
- Clinical
- Dysarthria
- Tremor
- Amyotrophy
- Frontal signs
- Levodopa-resistant parkinsonism
- Supranuclear palsy
- Corticobasal degeneration
- Pathology
- Spinal motor neurons: Loss
- Tau inclusions
- Genetics
- MAPT variant syndrome: Parkinson-ALS
197
- Nosology: Brait–Fahn–Schwarz disease
- Epidemiology: 1 patient
- Genetics
- Mutation: Pro494Leu; Heterozygous
- Clinical
- Onset age: 6th decade
- Parkinsonism
- ALS syndrome: After Parkinsonism
- Weakness
- Fasciculations
- Upper motor neuron: Spasticity; Tendon reflexes brisk
- Speech: Dysarthric
- Course: Progressive over 2 years
- Laboratory
- NCV: CMAP amplitude, small
- EMG: Denervation, diffuse
- Epidemiology: 1 family, 2 patients
- Genetics
- Mutation: Homozygous; Ser352Leu
- Inheritance: Recessive
- Clinical
- Onset age: 3rd decade
- Respiratory failure, Acute
- Learning difficulty: Early onset; 1 patient
- Pathology
- Tau accumulation: In some neuron cell bodies
● Signal transducer and activator of transcription 5B (STAT5B)
- Epidemiology: 2 patients, 1 family
- Genetics
- Mutation: Glu315Ala
- STAT5B protein
- Signal transduction
- Transcription activator
- Clinical
- Growth retardation: Short stature
- Dysmorphic face: Ptosis
- Motor neuropathy
● Autosomal Recessive
- Epidemiology: 1 patient
- Clinical
- Weakness: Bulbar; Arms; Respiratory failure
- Ophthalmoplegia: Reduced gaze excursion, especially up; Supranuclear
- Progression: Over 1 year
- Laboratory
- EEG: Diffuse slowing
- CT: Atrophy of frontal lobe & midbrain
- Brain: Abnormal anterior horn & corticospinal tracts; Bunina bodies
● RBM28
- Epidemiology: Arab Moslem kindred
- Genetics
- Mutation: Missense; Leu351Pro
- RBM28 protein
- Component of snRNP complexes
- Ubiquitous expression
- Regulates ribosome biogenesis
- Mutant: Ribosome depletion; Structural abnormalities of rough endoplasmic reticulum
- Clinical
- Skin
- Hair loss
- Widely varying severity
- Skin biopsy: Absence of mature hair follicles; Reduced β-catenin
- Pigment
- Flexural reticulate hyperpigmentation
- Facial pigmented nevi: Multiple
- Subcutaneous fat: Loss
- Hair loss
- Motor
- Progressive decline
- Combined upper & lower motor dysfunction
- Onset: 2nd decade
- CNS
- Mental retardation: Moderate to severe
- Endocrine: Central hypogonadotropic hypogonadism
- Delayed or absent puberty
- Central adrenal insufficiency
- Gynecomastia
- Skeletal
- Short stature
- Microcephaly
- Hypodontia
- Kyphoscoliosis
- Ulnar deviation of hands
- Skin
- MRI
- Hypoplastic pituitary gland with preserved hypothalamus
- Normal basal ganglia & white-matter
● SCN1A
- Nosology: Epileptic encephalopathy, early infantile, 6 (EIEE6)
- Epidemiology: Rare epilepsy syndrome
- Genetics
- SCN1A protein
- Clinical
- Ataxia: Gait
- Seizures: Severe; Onset 2 to 6 months; Pharmacoresistant
- Psychomotor delay: Moderate to severe
- Orthopedic
- Misalignment
- Knee hyperextension
- Foot: Pes valgus
- Crouching
- Movement disorders
- Onset age: Adulthood
- Lingual dyskinesia
- Rigidity
- Dystonic gait
- Treatable with levodopa
- Tendon reflexes: Reduced (30%)
- Laboratory
- EEG: Normal then Generalized spike-wave activity
- NCV: Normal
- EMG: Motor neuronopathy
130
- Chronic denervation, especially > 4 years
- Motor units: Prolonged; High amplitude; Polyphasic
● von Willebrand Factor A Domain-containing Protein 1 (VWA1)
- Epidemiology: 34 patients, 23 families
- Genetics
- Mutations
- Types: Loss-of-function; Duplication & Deletion
- Common in Europeans: G25Rfs*74 10 bp repeat expansion (3 copies vs 2 in controls)
- Allelic disorder: Hemifacial microsmia, Dominant
, Missense mutation
171
- Mutations
- VWA1 protein
- Location: Basement membranes; Extracellular matrix
- Interactions: Collagen VI; Perlecan; FGF pathway
- Secreted
- Clinical
- Onset age: 1 to 54 years
- Weakness
- Legs: Anterior > Posterior; Foot drop
- Thigh: Anterior & Posterior
- Arms: Mild or None
- Proximal & Distal
- Myalgia (40%)
- Sensory: Normal
- Tendon reflexes: Reduced in 50%
- CNS: Normal
- Skeletal: Pes cavus (90%); Joint contractures (50%)
- Course: Slow progression
- Young onset: Upper motor neuron signs; Gait disorder
- Laboratory
- Muscle pathology
- Fiber type groups
- Fiber sizes: Varied
- Ultrastructure: Mitochondrial paracrystalline inclusions
- EMG: Fibrillations, distal; Myopathic, proximal
- NCV: Motor axon loss
- Muscle MRI
- Uniform pattern
- Thigh: Vasti
- Legs: Gastrocnemius (Medial > Lateral); Peroneal
- Serum CK: Normal to 1,600
- Muscle pathology
Motor Neuron Disease: Mouse models
- Wobbler
52
● Vacuolar protein sorting protein 54 (Vps54)
- Mutation: Missense L967Q
- Motor neuron disease
- Spermiogenesis defect
- Neuromuscular degeneration (nmd): SMARD1
- Muscle deficient: Mouse chromosome19
- Onset: Hindlimb weakness
- Peripheral nerve: Axonal degeneration
- Neurons: Ventral horn & Brainstem motor; Vacuolar degeneration
- Death: 8 months
- Progressive motor neuronopathy (PMN)
28
- CNTF knockout: Mouse chromosome 19
- SOD transgenic
- Neurofilament heavy chain
: Overexpression
- PQBP-1 transgenic
35
- PQBP-1
- Transcription and/or mRNA processing factor
- Interacts with polyglutamine disease gene products
- Clinical: Progressive weakness with increasing age
- Pathology: Neuronal loss in spinal anterior horn; Loss of motor axons
- PQBP-1
Early description of SOD1 ALS family with A4V mutation

|

|

|
Patient information: Spinal muscular atrophy
Return to Neuromuscular Home Page
Return to Motor Syndromes
Return to Polyneuropathy Index
References
1. Neurology 1999;53:2187-2189
2. Human Mutation 2000;15:228-237, J Med Genet 2003;40:e39-e42
3. Neurology 2000;54:1534-1537
4. Am J Med Genet 2000;92:117-121; Am J Hum Genet 2010; Online March
5. Am J Med Genet 1998;75:193-195
6. Brain 2000;123:1612-1623, Nature Genetics March 2004
7. JNNP 1998;64:217-220
8. Human Mutation 2000;16:253-263
9. JAMA 2000;284:1664-1669
10. Acta Neuropathol 2000;100:603-607
11. Brain 2000;123:2160-2170
12. Brain 1992;115:1889-1900
13. Pediatr Neurol 2001;24:371-372
14. Pediatr Neurol 2001;25:59-61
15. Neuromuscular Disorders 1998;8:405-408
16. Nature Genetics 2001;29:160-165, Nature Genetics 2001;29:166-173
17. Am J Hum Genet 2002;70:January, Exp Neurol 2014;262 Pt B:91-101
18. J Neurosci 2001;21:9246-9254
19. Neuromuscular Disorders 2002;12:26-30
20. J Neurol 2002;249:290-293
21. Ann Neurol 2002;51;585-592
22. Brain 2002;125;1320-1325
23. Hum Mol Genet 2002;11;1605-1614
24. J Cell Biol 2001;152:1107-1114
25. American Journal of Medical Genetics 2002;110:301–307, Neurology 2022 Jul 14
26. Brain 2002;125:1624-1634, Neuropathol Appl Neurobiol 2024;50:e13013
27. J Neurochem 2002;82:1229-1238, J Neurol Sci 2006; Online January
28. Nature Genet 2002;On-Line October 21
29. Hum Mutation 2002;On-Line #552
30. J Mol Biol 2002;324:247–256
31. Ann Neurol 2002;52:680-683
32. Arch Neurol 2002;59:1921-1926
33. J Neurological Sci 2003;209:13-17
34. Nature Genet 2003;On-Line March 10, J Neuromuscul Dis 2025 Jul 15
35. Hum Molec Genet 2003;12:711–725
36. Am J Hum Genet 2003; Online June; Science 2009:323; 1205-1208 & 1208-1211, Neurology 2010; July 28, Neurobiol Aging 2015 Aug 15
37. Nature Genet 2003; Online June 29
38. Am J Hum Genet 2003; Online July
39. Muscle Nerve 2004;29:323–328, Neuromuscul Disord 2022;33:148-152, Mol Genet Metab 2023;138:107525
40. PNAS 2004
41. J Med Genet 2004;41:224–229, Neurology 2009;72:246–252
42. Neuron 2004;41:687-699
43. J Med Genet 2004;41:315–320, Am J Hum Genet 2004; Online September, Amyotroph Lateral Scler Frontotemporal Degener 2026 May 2
44. Am J Hum Genet 2004; Online April
45. J Peripher Nerv Syst 2004;9:122-123
46. Hum Mol Genet 2004;13:1677-1692
47. Am J Med Genet 2004; Online Sept
48. J Biol Chem 2004; Online Aug, Neurobiology of Aging 2010; Online Apr
49. Hum Mol Genet 2004;13:R195-R202
50. Nature Genet 2005; Online March
51. NeuroReport 2005;16:657-661
52. Nature Genetics 2005; Online October
53. Neurology 2005;65:1954–1957
54. Brain 2006; On line Feb 22, Neurology 2006; On line January 18, Neurology 2009;72:1669–1676
55. Nature Genetics 2006; Online Feb 26, Ann Neurol 2011;70:964–973
56. J Cell Biol 2006;172:733-745
57. Neurology 2006;67:120–124, Am J Hum Genet 2007; Online May, Eur J Neurol 2020 Nov 21, Ann Clin Transl Neurol 2020 Dec 4, J Peripher Nerv Syst 2026;31:e70099
58. Neurology 2006;67 On line June 28 , PLoS ONE 2010;5:29872
59. Hum Genet 2007 Mar 13, Brain 2022 Nov 16
60. Amyotroph Lateral Scler 2007;8:73-78
61. J Neurol Sci 2008 Feb 6
62. Ann Neurol 2008 Online Feb 20, Science 2008 Online Feb 27, Nat Genet 2008 Mar 30, Lancet Neurology 2008 Online Apr 5, Neurology 2012;78:1519–1526, Brain Commun 2026;8:fcag241
63. Am J Hum Genetics 2009;;84:85-88
64.Neuromuscul Disord 2009 Mar 20
65. Neurology 2009;72:1153–1159
66. Hum Mol Genet 2009;18:1288-300
67. Neuromuscul Disord 2009;19:193-195
68. Am J Human Genet 2009; Online July
69. Am J Human Genet 2009; Online August
70. Brain 2010 Online January
71. Neurology 2010;74:502-506, Cell Rep 2017;20:2100-2115
72. Am J Hum Genet 2010; Online February
73. PNAS 2010; Online April
74. Nature 2010;465:223-226, J Neurol Neurosurg Psychiatry 2011 May 25
75. Neurology 2010;75:539-546, Neurology 2012;78 On-line March
76. J Neurol Neurosurg Psychiatry 2010;81:572-577
77. Neurology 2010;75:611–618
78. Neuron 2010;67:575-587, Neurol Genet 2022;8:e200011
79. Neurology 2011;77:334-340, European Journal of Human Genetics 2012; Online April, Ann Neurol 2014; Online Nov
80. Neurobiology of Aging 2011; Online August
81. Ann Neurol 2011; Online August, J Peripher Nerv Syst 2022 Oct 12
82. Nature 2011; Aug 21, Brain 2024 May 4
83. Neuron 2011; Online September: A, B, Acta Neuropathol 2012; On-Line Jan, Brain 2012; Online Feb, Am J Human Genet 2013; Online February
84. PLoS One 2011;6(10):e26164
85. J Neurol Neurosurg Psychiatry 2011 Oct 25
86. PNAS 2011; On-line November
87. Annals Neurology 2011; On-line November
88. Hum Mol Genet 2012; Online Mar
89. Nature Genetics 2012; Online Apr
90. Brain 2012;135:1714-1723, American J Hum Genet 2013; Online May A, B, C, Brain 2014; Online Dec, Neurology 2016;87:2235-2243
91. Am J Human Genet 2012; On line June, Neuromuscul Disord 2022;32:806-810, Neurol Genet 2025;11:e200243
92. PNAS 2009;106:2794–2799, Braz J Vet Med 2026:48:e010525
93. Brain 2012; Online June 26, J Med Genet 2012; Online Dec, Metabolites 2025;15:491
94. Neurobiol Dis 2012; Online July
95. Nature 2012; Online July, Eur J Neurol 2022 Sep 29
96. Am J Human Genet 2012; Online Aug
97. Am J Human Genet 2012; Online Aug
98. Am J Human Genet 2012; Online Nov
99. Neurogenetics 2012; Online November
100. Mitochondrion 2013 Feb
101. Acta Neuropathol 2013;125:523–533
102. Acta Neuropathol 2013 May 15
103. Am J Human Genet 2013; Online May
104. Neurogenetics 2013 Aug 24, J Neurol 2014 Aug 23
105. Am J Human Genet 2013; Online Oct, Front Neurol 2022;13:865264
106. Am J Human Genet 2013; Online Oct
107. Nat Neurosci 2013 Nov 3
108. Acta Neuropathol 2014 Jan 3
109. Neurobiol Aging 2013 Dec 4
110. Neurology 2014; Online Feb
111. Neurology 2014, Genes (Basel) 2022;13(5)
112. Neurology 2014; Online May
113. Mol Genet Metab 2011;102:6-12
114. Nat Commun 2014;5:4287
115. Neuromuscular Disorders 2014: Online July
116. Neurology 2014;83:990-995
117. Neurology 2014; Online Oct, Eur J Neurol 2022;29:2056-2065
118. Am J Human Genetics 2014; Online October
119. Neuron 2014;84:324-331, medRxiv 2025 Jun 28
120. Science 2015; Online Feb, J Chin Med Assoc 2024;87:920-926
121. Nature Neuroscience 2013;16:851-855
122. Neurology 2015;84:2430-2437
123. Acta Neuropathol 2015 Sep 10
124. J Clinical Neuromuscular Disease 2015;17:69-71, Neurology 2016; Online June 8, Neurol Genet 2021;7:e599, Neuromuscul Disord 2022;32:527-532, J Peripher Nerv Syst 2026;31:e70145
125. Neurology 2015; Online December
126. Am J Human Genet 2016;98:473-489, Eur J Med Genet 2022 Jun 8
127. Parkinsonism Relat Disord 2016 Mar 3
128. PLoS One 2016;11(3):e0151376
129. Am J Hum Genet 2016; Online March, J Hum Genet 2022 Jan 28
130. Neurology 2016 Jun 17, Mol Neurobiol 2023 May 12
131. Amyotroph Lateral Scler Frontotemporal Degener 2016 Jun 27
132. Hum Mol Genet 2016;25:1559-1573, Ann Neurol 2019 Dec 3
133. Am J Hum Genet 2016; Online August
134. Am J Hum Genet 2016; Online September, Mol Genet Metab 2026;148:109873
135. Neuromuscular Disorders 2016: Online September
136. J Hum Genet 2016 Dec 8
137. JAMA Neurol 2017; Online Feb
138. J Neuromuscul Dis 2016;3:487-495
139. Brain 2017;140:1252-1266
140. J Neurol Neurosurg Psychiatry 2017 Apr 1, Acta Neuropathol Commun 2025;13:163
141. Sci Transl Med 2017;9(388), Front Neurol 2023:14:1086264
142. Sci Rep 2017;7:2116
143. Clin Genet 2018;93:301-309, Ann Neurol 2017 Dec 28
144. Neurol Genet 2017;3(4):e172
145. Neurobiol Aging 2017 Dec 27
146. Neurology 2018 Jan 19
147. J Neurol Neurosurg Psychiatry 2017;88:99-105
148. JIMD Rep 2017 Dec 7
149. Am J Hum Genet 2018 May 3
150. Hum Mol Genet 2016;25:2985-2996
151. Genet Med 2018 Mar 8
152. Neurology 2017;88:1226-1234
153. Neurobiol Aging 2019 Mar 11, Front Neurol 2024:15:1284459
154. Nat Commun 2019;10:1777
155. Brain 2019 Jul 23, Mol Biol Rep 2024;51:580
156. J Hum Genet 2019 Aug 17
157. Neurology 2019 Oct 2
158. Cell Rep 2019;26:2298-2306
159. Ann Neurol 2020 Jan 19, J Neurol 2020 Sep 30
160. Neuromuscul Disord 2020 Jan 17
161. Neuromuscular Disord 2020; Feb, J Hum Genet 2021 Mar 20, J Hum Genet 2024 Nov 15, Eur J Hum Genet 2025 Jun 4
162. Brain 2020 Mar 18
163. Front Neurosci 2020 Apr 28;14:316
164. J Clin Med 2020;9:E2222, J Neurol Neurosurg Psychiatry 2026 Jan 30
165. Amyotroph Lateral Scler Frontotemporal Degener 2020;30;1-8, Genes (Basel) 2021;12:1935
166. Neuron 2020 Nov 25:S0896-6273(20)30883-7, Neuron 2021;109:1945-1946
167. Eur J Neurol 2020 Dec 28
168. Int J Biochem Cell Biol 2020 Jun;123:105746
169. Orphanet J Rare Dis 2021;16(1):10
170. Brain 2021 Jan 18, Brain 2021 Jan 18
171. Front Cell Dev Biol 2020 Sep 9;8:571004
172. J Mol Neurosci 2021 Jan 22
173. Orphanet J Rare Dis 2021 May 7;16:207
174. Ann Neurol 2021 May 28, Ann Clin Transl Neurol 2022 Sep 22, Neuromuscul Disord 2025;53:105443
175. Neurol Genet 2021;7:e598
176. Neurobiol Aging 2021 Jun 1
177. Sci Rep 2021;11:13613, Sci Rep 2021;11:11868
178. Semin Pediatr Neurol 2021;37:100878
179. J Neurol Neurosurg Psychiatry 2021 Sep 13, Front Genet 2023;14:1208673
180. J Biol Chem 2021;101299
181. Front Genet 2021;12:746060
182. J Child Neurol 2020;35:717-723
183. Sci Rep 2020;10:20738
184. N Engl J Med 2020;383(2):109-119, J Neurol 2024 Jun 3
185. Am J Hum Genet 2022 Jan 27, Mol Genet Genomic Med 2023;11:e2131
186. NPJ Genom Med 2022;7:8
187. Hum Mutat 2022 Jul 11
188. J Med Genet 2022 Jul 25
189. Front Neurol 2022 Aug 3;13:939775
190. Sci Rep 2022;12:14739
191. Genet Med 2022 Nov 18
192. Brain 2023 Feb 17
193. Acta Neuropathol 2023 Mar 31, Brain 2023 Dec 11
194. Cells 2023;12:847
195. Hum Mol Genet 2023 Apr 18
196. Molecules 2023;28:5801; Rev Neurosci 2023 Aug 2, Science 2026 Feb 5
197. Neurol Sci 2023 Sep 20
198. Front Neurol 2023;14:1242472
199. RMD Open 2023;9:e003431
200. J Med Genet 2023 Oct 27
201. Mov Disord 2022;37:384-391, Ann Med Surg (Lond) 2022;84:104840
202. Neuromuscul Disord 2024:34:114-122
203. Neurology 2009; 72:1634–1639
204. Brain 2024 Jun 17
205. J Med Genet 2024 Feb 27
206. Neurology 2002 ;59:770-772
207. Ann Neurol 2024;95:596-606
208. Muscle Nerve 2024;70:257-264
209. J Neurol Neurosurg Psychiatry 2024 Jul 2
210. EMBO Rep 2024 Jul 18
211. Brain 2024 Sep 23
212. Ann Neurol 2024 Oct 23, Am J Med Genet A 2026 Feb 26
213. Sci Rep 2024;14:29880
214. Orphanet J Rare Dis 2024;19:476
215. Acta Neuropathol Commun 2025;13:2
216. J Neurol 2025;272:181
217. Alzheimers Dement 2025;21:e14566
218. J Neurol 2025;272:271
219. medRxiv 2025
220. J Neurol 2025;272:191
221. Int J Mol Sci 2025;26:2905
222. Orphanet J Rare Dis 2025;20:209
223. Ann Clin Transl Neurol 2023;10:408-425
224. Neuromuscul Disord 2025:53:105445
225. JAMA Netw Open 2025;8:e2536348
226. J Clin Invest 2025:e184474
227. Neurol Neurochir Pol 2025 Dec 3
228. Brain 2026 Feb 12
229. J Clin Endocrinol Metab 2025;110:e2485-e2497
230. Muscle Nerve 2026 Jun 9
231. JAMA Neurol 2026 Jul 6
232. Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration 2025;26:573–590
7/16/2026