|
Home, Search, Index, Links, Pathology, Molecules, Syndromes, Muscle, NMJ, Nerve, Spinal, Ataxia, Antibody & Biopsy, Patient Info |
Motor Syndromes
Motor Neuron Disorders: Differential Diagnoses
|
|
||||||||

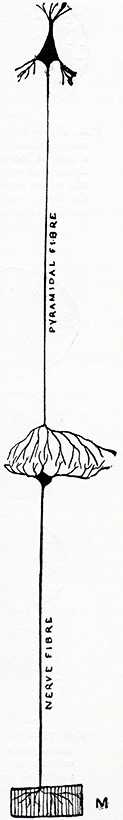

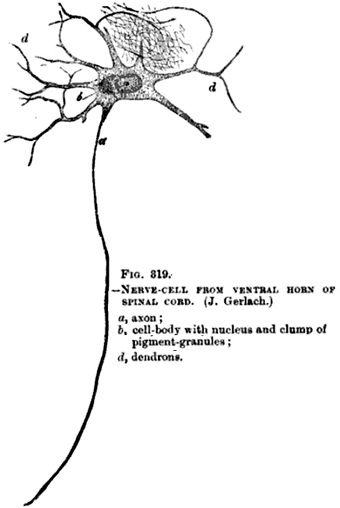
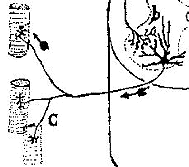
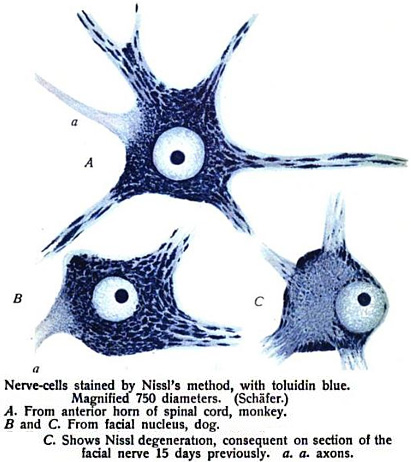
Paraneoplastic Motor Neuropathy 25
|
Paraneoplastic Lower Motor Neuron Syndrome 8, 25
- Epidemiology: Single patient, 72 year old female
- Onset: 4 months before diagnosis of tumor
- Clinical
- Weakness
- Asymmetric at onset
- Arms & Legs
- Severe
- Lower motor neuron only
- Bulbar: Hypophonia; Dysphagia; Unilateral facial paresis
- Painless
- Course
- Progressive over months
- Improvement after tumor removal
- Long-term residual disability
- Sensation: Normal
- Tendon reflexes: Absent
- CNS: Transient dizziness & Nystagmus
- Weakness
- Associated with
- Ductal adenocarcinoma of breast
- Laboratory
- Antibodies
- Serum binding to βIV spectrin, isoform I (Bands at MW 250kD & 140kD)
- Serm binding to axon initial segments & nodes of Ranvier in rat brain
- Electrophysiology
- EMG: Denervation
- NCV: Small CMAPs; No conduction block
- MRI: Spinal cord with high signal spots on T2
- ? Neuronopathy
- Antibodies
- Immunosuppressive treatment: No response
- Also see: Paraneoplastic Motor Neuropathy
HOPKINS' SYNDROME: Acute Post-asthmatic Amyotrophy 29
- Clinical
- Onset
- Age: 1 to 13 years
- After acute asthmatic attack: Latency 1 to 18 days
- Mild pain: Limb, neck or meningismus
- Rapid onset weakness
- Weakness
- Single limb; Asymmetric; May be Proximal > Distal
- Severity: Mild to Severe
- Arm or Leg
- Wasting: Affected limb
- Sensory: Normal
- Tendon reflexes: Reduced
- Meningismus: Some patients
- Course: Monophasic; May recur with repeat asthma attack
- Prognosis: Permanent paralysis
- Onset
- Laboratory
- EMG: Denervation
- NCV in limbs
- CMAPs: Reduced or Absent, Asymmetric
- SNAPs: Preserved
- CSF
- Pleocytosis
- Protein: ± Increased or Normal
- MRI: May show signal (T2) in
- Spinal cord
- Anterior roots
Post-Polio Syndrome
|
|
Monomelic Amyotrophy

- Nosology
- Hirayama disease
- Course: Progressive weakness over 1 to 4 years; Then plateau
- More related to flexion myelopathy
- O'Sullivan-McLeod syndrome
- Course: Slow progression over decades
- Juvenile muscular atrophy of distal upper extremities (JMADUE)
- Hirayama disease
- Epidemiology
- Possible etiologies
- Cervical myelopathy, central
- Neck flexion induced
- Forward displacement of posterior cervical dural sac
- Increased segmental & range of motion in cervical spine
- Cervical cord & vessel compression
- Tightness within cervical dura
- Due to: Differential elongation of spinal cord & dura during puberty
- Anterior horn of spinal cord: Atropy & Ischemic changes
- Neck flexion induced
- Motor neuron disease
- Genetic associations
- Cervical myelopathy, central
- Clinical
17
- Onset
- Age
- Mean 15 to 20 years; Range 13 to 25 years
- Up to 40 years in India
- Females: 2 years younger than males 26
- Weakness: Distal; Single limb
- Age
- Weakness
- Asymmetric
- Often predominant in one arm
- Subclinical involvement of other arm: Common
- Locations
- Distal predominant (97%): Proximal (Up to C5) > Distal in 3% to 10%
- Hand & Forearm
- C8 & T1 ± C7, innervated muscles
- Side: Right 1x to 3x > Left
- Cold paresis: More weakness & stiffness in cold
- Asymmetric
- Atrophy
- Hands
- "Oblique amyotrophy"
- Wasted: C7 muscles
- Preserved: Brachioradialis belly (C6)
- Tremor or Minipolymyoclonus (80%)
- On finger extension
- Irregular & Coarse
- Occasional other features
- Weakness: Other
- Ipsilateral shoulder
- Progression to opposite limb
- Frequency: 18% - 40%
- Latency: Range 2 to 120 months; Mean 43 months
- Usually milder weakness than 1st limb
- Worsening on exposure to cold
- Movements
- Fasciculations
- On affected side (47% to 66%)
- May not be symptomatic
- Contractile fasciculation
- Minipolymyoclonus (85%)
- Fasciculations
- Sensory loss
- Mild or none in affected limb
- Rarely prominent
- Subjective numbness (20%)
- Pin loss (C8 - T1)
- Discomfort
- Cramps & Spasms (30%)
- Neck pain
- Autonomic: Hyperhidrosis (18%)
- Weakness: Other
- Tendon reflexes: Usually normal or reduced; Occasionally increased
- No cranial nerve, leg or pyramidal changes
- Disease course
- Possible treatments
- Neck bracing: Hard collar
- Surgical
23
- Cervical spine stabilization
- Better prognosis with
- Younger age at surgery: Mean 19 years
- Shorter disease duration: Mean 22 months
- No: Babinski sign or Increased DTRs
- Surgery type
- Anterior only approach: Likely sufficient
- Stabilization only: Better than decompression
- Onset
- Laboratory
- EMG
- Location of pathology
- Most common: C8-T1
- Partial involvement: C7
- Spared: C5-C6
- Occasional: Legs
- May be present in asymptomatic limbs
- Acute & ongoing denervation in 45% to 70%: Fibrillations
- Chronic denervation
- In affected limb(s) (100%): Especially fasciculations
- Opposite arm or lower extremities: 30% to 100%
- Location of pathology
- NCV
- CMAPs: Small in affected limbs; Especially ulnar nerve
- Velocities: Normal
- Sensory: Normal
- Sympathetic skin response: May be abnormal
- MRI
27
- Cervical pathology
- Spinal cord atrophy
- Frequency: 30% to 50%
- Locations: C5 to C7
- May be asymmetric
- Anterior horns, gray matter: T2 signal at C5-C7
- Cervical lordosis: Reduced
- Neck flexion
- Posterior epidural venous plexus: Engorgement
- Posterior dura: Anterior shifting
- Post-gadolinium T1 enhancement of posterior epidural plexus
- Epidural flow voids
- ? Some patients with inelastic dura
- Spinal cord compression with neck flexion 1
- Spinal cord atrophy
- Other studies
3
- No major spinal anomalies
- Mild flexion-induced cord displacement
- Cervical pathology
- EMG
- Differential diagnosis: HMN & Distal SMA, especially HMN Type 5
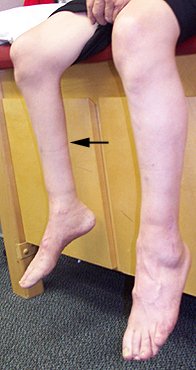
Amyotrophy: Benign Lower Extremity (Crural) 11
- Epidemiology
- 70 cases
- Male > Female: 5:1
- Onset age: Mean 20 to 41 years
- Clinical
- Weakness: Posterior legs
- Usual: Posterior calf; Peronei; Hamstrings
- Rare: Quads
- Often bilateral (50%)
- Clinical or Subclinical
- Asymmetry: Some
- No sensory, bulbar or upper motor neuron signs
- Tendon reflexes: Most often reduced
- Course
- Insidious onset
- Slow progression: Few years
- Stabilization
- Weakness: Posterior legs
- Laboratory
- MRI
- Loss of muscle with fat replacement
- Distribution: Lower extremities
- Distal leg: Posterior compartment
- Thigh: Long head of biceps
- EMG: Denervation
- Muscle biopsy: Denervation; Grouped atrophy
- MRI
| Benign lower limb amyotrophy: MRI | |
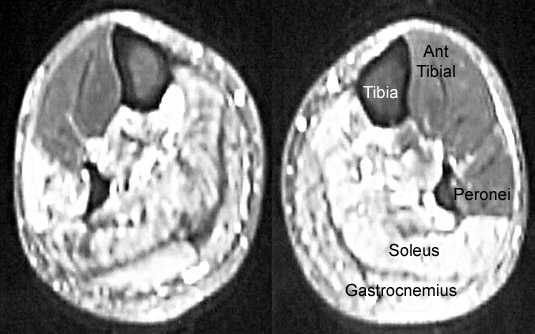 From: M Al-Lozi Legs: Involvement of posterior muscle groups
|
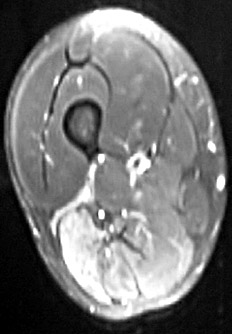 From: M Al-Lozi Thigh: Long head of biceps involved
|
MND: Finger Extension Weakness + DOwnbeat Nystagmus (FEWDON-MND) 22
- Epidemiology: 14 patients; Sporadic
- Clinical
- Onset age: 2nd to 4th decade
- Motor Neuron Disease
- Weakness
- Onset: Finger extension, unilateral
- Asymmetric
- Finger drop, bilateral
- Distal ≥ Proximal muscles
- Bulbar & Respiratory: Normal
- Progression
- Course: Slow over years
- Legs: Weakness later in course
- Fasciculations (70%)
- Tendon reflexes: Present; Often brisk
- Upper motor neuron
- Tone mildly increased in some
- Plantar reflex: Normal; Flexor
- Weakness
- Eye
- Nystagmus
- Downbeat: Later in disease course
- Gaze evoked
- Oscillopsia
- Diplopia
- Skew Deviation
- Esophoria: Comitant
- Saccadic dysmetria
- Nystagmus
- Sensation: Normal
- Laboratory
- NCV: Motor axon loss
- EMG
- Denervation & Reinnervation
- Chronic > Acute
- Motor units: Large
- CSF: Normal
- Brain MRI: Normal
"Primary Muscular Atrophy" (PMA) 15
|
|

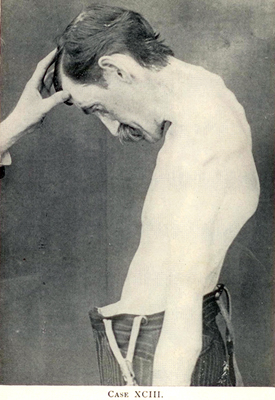
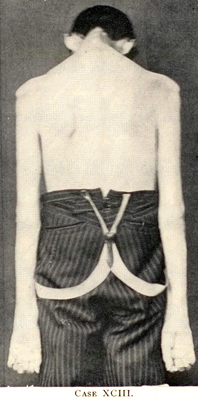
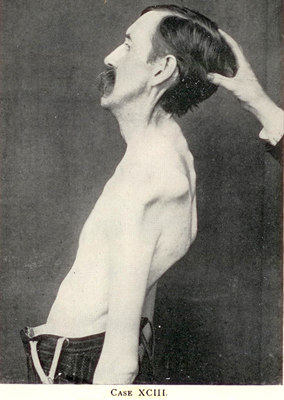
Primary Lateral Sclerosis (PLS) 2
|
Dominant, Adult onset 4p16 c9orf72 FIG4: 6q21 SPG7 PLS + FTD PSEN1: 14q24 TBK1: 12q14 Recessive Childhood onset ALS2 ERLIN2 SIGMAR1 SPG11 Adult onset ATXN2 Sporadic, Adult onset Mills (Asymmetric) syndrome 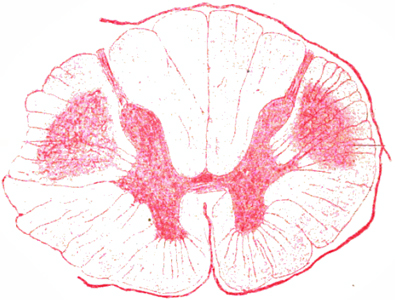 From Bramwell: Diseases of the Spinal Cord PLS: "Sclerosis" of pyramidal tracts
(Upper thoracic cord) |
 "About the Case of primary systematic Degeneration of the
Pyramidal tracts with Symptoms of a general spastic Paralysis"  Deutsche Zeitschrift für Nervenheilkunde 1894;5:225–246 |
PLS
- Nosology
- Pure upper motor neuron disease/dysfunction (PUMND)
- ? Discrete syndrome vs ALS variant
- Variant: Mills (Asymmetric) syndrome
- Definition
- Progressive upper motor neuron dysfunction,
- No: Lower motor neuron signs; Hereditary spastic paraplegia
- Frequently involves: Limbs & bulbar regions
- History: Early description by Adolf von Strümpell (1853-1925) 19
- Epidemiology
- 1% to 5% of sporadic motor neuron disease
- Male = Female
- Genetics
- Clinical
- Onset
- Spasticity
- Legs before Arms
- Slow progression
- Age: Most common in 5th & 6th decade; Range 34 to 78 years
- Spasticity
- Motor dysfunction
- Corticospinal ± Corticobulbar tract dysfunction
- General patterns
- Paraparetic: Legs > Arms
- Uncommon
- Earlier age onset
- Rule out HSP
- Bulbar
- Asymmetric: Mills syndrome
- Rule out
- Extrapyramidal disease
- Neurodegenerative disorder
- Paraparetic: Legs > Arms
- Spasticity
- Legs > Arms
- Bulbar: Dysarthria; Dysphagia
- Especially with disease onset > 45 years
- May be selectively present
- Pattern: Flexors in arms, Extensors in legs
- Tendon reflexes: Brisk
- Plantar reflex: Normal or upgoing
- Pseudobulbar Affect: Emotional lability
- Treatment
- Spasticity & Stiffness: Baclofen; Levodopa
- Pseudobulbar affect
- Dextromethorphan & Quinidine
- Tricyclic antidepressants
- General patterns
- Symmetry
- Symmetric: 20% to 50%
- Asymmetric: 50% to 80%; May be classified as Mills syndrome
- No lower motor neuron change
- Respiratory
- Vital capacity reduced with bulbar involvement
- Rarely symptomatic
- Corticospinal ± Corticobulbar tract dysfunction
- Sensory: Normal
- Cortical
- Frontal lobe dysfunction (10% to 20%): Mild
- Cognition impaired (36%): Mild; Language & Verbal fluency
- EOM disorders (25%): Saccadic pursuit or Up gaze reduced
- Bladder dysfunction
- Frequency: 30% to 50%
- Urgency
- Normal until later (Mean 4.5 years) in disease course
- Progression
- Gradual & Slow
- May stabilize after several years
- > 3 years; Up to 3 decades
- Lower motor neuron signs
- Frequency: 40%
- Onset: 3 to 10 years after onset
- Evolution to ALS after 4 years
- Rarely
- 20% develop denervation on EMG
- Differences from ALS: PLS has less
- Bulbar onset
- Weakness
- Focal
- Limb
- Fasciculations
- Weight loss
- Vital capacity reduction
- Speed of progression
- Onset
- Laboratory
- Magnetic stimulation
- Absent or prolonged cortical motor evoked latencies
- MRI
24
- Motor cortex: Medial
- Precentral gyrus, Focal atrophy
- Corpus callosum
- White matter: Corticospinasl tract
- Cerebellum: White matter; Body; Splenium
- Other: Insular, Inferior frontal; Left pars opercularis
- PET & FDG-PET: "Stripe sign"
- Reduced glucose consumption in pericentral region
- Hypometabolism in primary motor cortex
- Also occurs in ALS
- fMRI: Cerebro-cerebellar connectivity increased
- Central motor conduction times: Prolonged
- EMG
- Few fibrillations or fasciculations in 1 or 2 muscles
- Motor units: Normal or Mildly reduced number
- Normal: Serum; CSF; Spinal cord imaging; Serum CK
- Magnetic stimulation
- Disease association: ? Breast cnacer
- Pathology
- Corticospinal tract: Axon loss
- Normal: Anterior horn cells ± Betz cells
- Differential diagnosis
- Structural disorders
- Spinal
- Foramen magnum
- Hydrocephalus
- Hereditary spinal disorders
- Systemic: Infections & Toxins
- "Degenerative"
- ALS
- Progressive supranuclear palsy
- Structural disorders
- PLS variant: Mills syndrome (PLS + Hemiparesis, Progressive)
20

- Epidemiology: Subgroup of patients with PLS syndromes
- Clinical
- Onset
- Age: 19 to 88 years
- Regions: Arm, Leg or Bulbar
- Spastic hemiparesis
- May begin in leg or arm
- Tone: Increased; Spastic
- Distribution: More severe
- Corticospinal pattern
- Arm flexion
- Leg extension
- Tendon reflexes: Increased; Asymmetric
- Plantar response: Extensor on hemiparetic side
- Progressive: May plateau
- Sensory: Normal
- Course
- Progressive over years
- May develop
- Bulbar signs (70%): Onset after or before hemiparesis
- Contralateral signs (40%)
- Onset
- Laboratory
- EMG: Normal
- Brain MRI: Normal or Cortical atrophy, mild
- Brain PET: Motor & Premotor areas
- Hypometabolism: Contralateral > Ipsilateral to weakness
Pseudobulbar Paralysis, Selective
|
|

PLS: Recessive, Juvenile onset
● Alsin (ALS2)
- Epidemiology: Cypriot family
- Genetics
- Mutation
- Deletion: In Exon 9
- Homozygous c.2980-2A>G mutation at splice acceptor site of intron 17: Frameshift
- Allelic with
- ALS2: Mutations in another region of alsin gene
- Familial spastic paraparesis, infantile onset (IAHSP)
- Mutation
- Clinical
- Onset: Childhood
- Spasticity: Bulbar; Extremities
- Gaze paresis: Saccadic
- Normal: Cognition; Sensation
- Progression: Slow; Some remain ambulatory for decades
- Laboratory
- Central motor conduction times: Delayed or Unrecordable
- EMG: No denervation
- Brain imaging: Normal
PLS: Dominant, Adult onset (PLSA1)
● Chromosome 4p16; Dominant
- Epidemiology
- Single French-Canadian family
- Male:Female = 1:1
- Onset
- Age: 30 to 60 years
- Spasticity: Leg; Asymmetric
- Clinical
- Spasticity: Arms & Legs; Asymmetric
- Strength: Normal
- Reflexes
- Tendon: Diffusely increased
- Plantar: Extensor
- Dysphagia: 60%
- Sensation: Normal
- Laboratory
- EMG: Denervation, mild, distal, chronic
- MRI: Normal or Spinal cord atrophy, mild
PLS: Dominant, Adult onset 28
● Presenilin 1 (PSEN1)
- Epidemiology: 1 family, 3 patients
- Genetics
- Mutation: Pro284Leu
- Allelic disorders
- Acne inversa, familial
- Alzheimer disease, type 3 ± Spastic paraparesis & Apraxia
- Cardiomyopathy, dilated, 1U
- Dementia, frontotemporal
- Pick disease
- Acne inversa, familial
- PSEN1 protein
- Catalytic component of γ-secretase.
- γ-secretase;: Proteolytic cleavage of APP
& NOTCH receptor proteins
- Clinical
- Onset age: 27 to 37 years
- Pyramidal
- Spastic paraparesis/quadriparesis: Asymmetric
- Gait disorder
- Dysarthria
- Pseudobulbar affect
- Tendon reflexes: Brisk
- Face: Paresis
- Cognitive disorders
- Urinary incontinence
- Course: Progressive
- Laboratory
- CNS: Corticospinal tract microgliosis; Aβ pathology in motor cortex
- EMG: Normal
- Brain MRI: Atrophy, Global + Cervical spine
PLS with Frontotemporal Dementia 12
● Sporadic
- Epidemiology: 2 patients
- Differential diagnosis
- Onset
- Age: 6th to 8th decade
- Dementia
- Dysarthria
- Clinical
- Cortical
- Dementia
- Aphasia
- Motor
- Spasticity
- Bulbar dysfunction: 1 patient
- Strength: Normal
- Tendon reflexes: Brisk
- Plantar responses: Extensor
- Course
- Slow progression
- Death after 7 years
- Cortical
- Pathology
- Neuronal loss: Frontotemporal & Motor cortex
- Inclusions: Ubiquitin & TDP-43 positive cytoplasmic
Return to Polyneuropathy Index
References
1. Spine 1997;22:486-492
2. J Neurol Sci 1999;170:5-10, JNNP 2001;71:615-620, Neurol Clin 2015;33:749-760, Acta Neurol Scand 2016 Nov 15
3. J Neurol 1999;246:1069-1074
4. Clin Neurol Neurosurg 2000;102:109-112
5. JNNP 2000;69:257-261
6. J Neurol 2000;247:654-655
7. Acta Neurol Scand 1997;96:14-21
8. Neurology 1999;53:852-855, PNAS 2001;98:6945-6950
9. Nature Genetics 2001;29:160-165, Neuropediatrics 1995;26:313-319, Neurology 2009; Online January
10. Acta Neurol Scand 2003;107:215–220, Muscle Nerve 2008 Feb 20
11. J Neurol Sci 1994;107:153-161, Acta Neurol Scand 1992;85-397-400
12. Neurology 2007;69:1800-1801
13. Neurologist 2009; 15:156-160
14. Neurology 2009;72:1948–1952, Muscle Nerve 2012; Online August, Neurology 2021 Feb 26
15. Neurology 2009;73:1686–1692
16. Neuromusc Disord 2012;22:394-400
17. J Child Neurol 2011;26:1542-1547
18. Neurology 1990;40:884-886
19. J Neurol 2012;259:2211-2220
20. Case Rep Neurol 2013;7:191-195
21. Muscle Nerve 2016 May 3
22. Muscle Nerve 2017 Apr, Muscle Nerve 2025 Jan 23
23. World Neurosurg 2017 May 27, World Neurosurg 2023 Jan 6
24. J Neurol 2019 Jul 19
25. J Neuropathol Exp Neurol 2021 Aug 17
26. Neurologia (Engl Ed) 2022 Aug 10
27. J Neuroimaging 2022;32:596-603
28. Alzheimers Dement 2024 Jul 27
29. Pediatr Neurol 2025:163:4-6
1/24/2025