Home, Search, Index, Links, Pathology, Molecules, Syndromes,
Muscle, NMJ, Nerve, Spinal, Ataxia, Antibody & Biopsy, Patient Info
Acute Immune Polyneuropathies
|
"Classic" Guillain-Barré Associated disorders Childhood GBS Clinical features Electrodiagnostic features Epidemiology Laboratory features Morbidity Pathology Prognosis Prodrome Therapy Acute Immune Neuropathies Autonomic Motor (AMAN) Sensory Cranial nerve variants Ataxia & Ophthalmoplegia Bickerstaff Facial diplegia Miller Fisher Syndrome Pharyngo-Cervico-Brachial IgM vs GalNAc-GD1a Pathology General topics Acute immune neuropathies Antibodies Classification Differential diagnosis General Principles GBS-like syndromes: Treatment |
 Georges Guillain |
  Ross & Bury 1893 |
Classification of Acute Immune Neuropathies
- Demyelinating (± 2° axonal loss)
- Motor + Sensory
- Classic Guillain-Barré
- CIDP with acute onset: More prominent demyelination on NCV
- Motor
- Motor + Sensory
- Axonal
- Motor + Sensory (AMSAN)
- GBS-like syndrome
- Vasculitis
- Motor: Associated with
- Prodromal infection: Campylobacter jejuni; Haemophilus influenzae
- Serum antibodies: IgG vs GM1, GM1b, GD1a or GalNAc-GD1a ganglioside
- Sensory
- Autonomic
- Motor + Sensory (AMSAN)
- Cranial Nerve Syndromes: Associated with
- Campylobacter jejuni prodrome
- Serum IgG vs GQ1b ganglioside
- Miller-Fisher
- Facial diplegia
- Bickerstaff's Brainstem encephalitis
- Pharyngo-Cervico-Brachial
- Other GBS variants with autoantibodies
Acute immune neuropathies: General principles
|
Acute immune neuropathies have several features Common features Vary & Distinctive for particular syndromes Suggest a Different diagnosis Acute motor dysfunction Treatments (Subjective) |
- Epidemiology
- Incidence
- Europe & North America
- 0·8–1·9 (median 1·1) cases/100,000 people per year
- Age association
- Childhood: 0.6/100,000 people per year
- Elderly: 2.7/100,000 people per year
- Europe & North America
- Males slightly > Females
- Incidence
- Common features
- Prodromal illness
- 50% of patients in 2 weeks prior to disease onset
- Respiratory or GI
- Strongest association: C. jejuni
- GBS syndrome peak frequencies
28
- Correlate with hospitalizations for Pneumonia & influenza
- No significant association with vaccinations
- 50% of patients in 2 weeks prior to disease onset
- Progression
- Average: 5 to 10 days
- Spectrum: 2 to 28 days
- Course
- Usually monophasic
- Rare relapses
- Prognosis
- Recovery in most
- Cerebrospinal Fluid (CSF)
- High protein (> 0.55g/L)
- Few or no cells
- Prodromal illness
- Variable features
- Motor, Sensory, or Autonomic involvement
- Tendon reflexes: More frequently preserved or increased with motor predominant syndromes
- Demyelinating vs. Axonal Pathology
- Degree of CNS involvement
- Humoral vs. Cellular immunity
- Features suggesting another diagnosis
- Sensory level: Spinal cord syndrome
- Severe bladder or bowel dysfunction: Spinal cord syndrome
- Marked asymmetry: Vasculitis
- > 50 WBC/mm3
- Very slow nerve conduction velocities (< 32 M/s):
CIDP
- Relapses, or a chronic course, may be more likely
- Differential diagnosis of acute motor dysfunction
- Treatment of GBS-like syndromes: Plasma exchange vs IV Ig
- Overall: No difference in efficacy
- Indications for rapid treatment: 1st 2 weeks of disease
- Bulbar disorders
- Respiratory dysfunction
- Inability to walk without assistance
- Probably indicated: Milder weakness; Early in disease course
- IV Ig: ? Primary therapy; 2 gm/kg total over 2 to 5 days
- Slightly lower cost
- Slightly fewer side effects
- Easier to administer
- Use in
- Children
- Rural settings with no access to plasma exchange
- Syndromes with anti-glycolipid antibodies
- Pure motor syndrome (IgG vs GM1)
- Miller-Fisher syndrome (IgG vs GQ1b)
- Patients with
- Diarrhea prodrome
- Infectious disorders (HIV)
- Autonomic instability
- Poor venous access
- Relapses of weakness
- Plasma Exchange: 4 or 5 treatments over 7 to 10 days
- ? Fewer late relapses
- No allergic reactions
- Efficacy somewhat better documented
- Use in
- Adults with good venous access
- Very recent onset of symptoms (1 to 3 days)
- History of side effects with IV Ig
- Pregnancy
- Congestive heart failure
- Renal insufficiency
- IgA deficiency
"Classic" Guillain-Barré Syndrome
|
Autoantibodies Clinical features Electrodiagnostic Laboratory Morbidity Other features Pathology Prodrome Prognosis Therapy |
 English translation: By Bassam Malo MD |
- Epidemiology
- Incidence: 1 to 2/100,000/year
- Male: Female = 1.25: 1
- Peak ages: Bimodal
- Children & Young adults
- > 55 years
- Zika infection: Colombia
- Winter month peak: United States, Australia & Asia
- Genetic risk factors
38
- Familial cases

- Reported associations with occurrence, prognosis & severity of GBS
- Conflicting results
- FcγRIIa-H131
allele homozygosity (vs R131)
4
- More common than in healthy controls
- Higher risk for severe disease than other genotypes
- Same allele protective against lupus nephritis
- Familial cases
- GBS Prodrome
- Upper respiratory: + CMV titers = 18%
- Younger patients
- More sensory loss & cranial nerve involvement
- More severe disease
- Respiratory insufficiency more common (65%)
- Longer median time until independent locomotion
- Antibodies
- Higher Frequency of serum IgM vs GM2 ganglioside
- Also see IgM vs GalNAc-GD1a ganglioside
- Gastrointestinal: + Campylobacter jejuni titers = 28%
- Motor predominant
- More severe outcome
- + Campylobacter titers in US GBS patients overestimate prevalence of infection
- Mycoplasma pneumonia
- Associated with antibodies to galactocerebroside (GalC)
- Frequency: 5% of GBS patients in Japan
- ? Associated with younger patients
- Other associated disorders
- Other infections
- Epstein-Barr virus
- HIV
- Dengue fever: 6x risk
- Zika virus
- Frequency: 8x to 20x risk
- More severe disease: More functional disability & respirator use
- More EOM involvement
- Prodrome features, common: Rash; Conjunctival hyperemia; Retro-ocular pain; Pruritis
- GBS types: Demyelinating, AMAN, Transient polyneuritis
- Chikungunya
33
- Epidemiology
- Seasonal association
- India: 16% of GBS
- No statistical association in Mexico
- GBS type: Demyelination in 75%
- Epidemiology
- COVID-19
- Japanese encephalitis
- Hospital diagnosed infections & antibiotic use
- GBS frequency: OR 13.7x higher (4.3% vs 0.3%) in Denmark 35
- Time: More within 1 to 2 months of infection
- Infection types: Lower respiratory, GI, Septicemia
- ? Hepatitis A
- ? Oropouche virus (OROV): No effect on GBS phenotype 44
- Vaccinations
- Tetanus toxoid; Influenza; ± Polio (oral)
- Rabies
- Vaccines: Myelin-containing (Semple); Suckling mouse brain
- Usually > 10 years old
- Some cases associated with sensitization to myelin basic protein
- Occurs in clusters
- Post-partum: 1st 2 weeks with higher risk
- Surgery
- ? Graft vs Host disease
- Drugs
- Upper respiratory: + CMV titers = 18%
- Clinical features
- Onset
- Weakness: Most often symptomatic in legs
- Pain: Low back & legs
- Paresthesias: Distal
- Weakness
- Distribution: Proximal + Distal; Symmetric
- Severity: Quadriplegia in 30%; Bedbound another 30%
- Respiratory failure
- Vital capacity < 1 liter: Observation in ICU necessary
- ~33% of GBS require intubation
- Indications for intubation
- Vital capacity < 12 to 15 ml/kg: Especially with rapid decline
- Negative inspiratory force (NIF) < 25 cm H2O
- Hypoxemia: PaO2 < 80 mm Hg
- Difficulty with secretions
- Time of onset: 7 days
- Time on respirator: 50% < 3 weeks
- Usually 2° to muscle weakness
- Occasionallly related to aspiration
- Cranial Nerves (70%)
- VII
- Symmetric: Occurs early in parallel with weakness
- Asymmetric
- Occurs later in disease course
- Other weakness may be stable or improving
- Extra-ocular: Overlap with Miller-Fisher
- Tongue: Symmetric; Common (50%)
- VII
- Sensory
- Paraesthesias: Initial symptom in 50%; Eventually occur in 70% to 90%
- Pain
23
- Prominent in 70%
- Location: Extremities > Trunk
- Timing: May occur with prodrome (35%), disease (65%) or sequelae (40%)
- Associations
- Neuropathy: In back, hips & legs at onset; Myalgias; Occasional radicular
- Immobility: Myalgias
- More severe disease
- More prominent sensory involvement
- Recovery phase: Distal; Legs > Hands; Dysesthesias
- Loss
- Distal; Symmetric
- All modalities involved
- Tendon reflex loss
- Early in most (70%) but not all patients
- Progressive reduction during 1st week
- Distribution: Ankles most frequently lost; Biceps most frequently spared
- Associations: Sensory loss; Weakest limbs; Distal
- Spared reflexes all during disease course suggests another diagnosis
- Autonomic dysfunction
- Frequency: 60%
- More common in more severe syndromes
- Blood pressure
- Transient hypertension or, less often, hypotension
- Increased sensitivity to anti-hypertensive medications
- Cardiac arrhythmias: Sinus tachycardia; Bradycardia
- Bladder: Urinary retention; Sphincter symptoms in 10% to 15%
- GI: Ileus
- Test: Bilateral ocular pressure x 25 sec; Produces bradycardia (< 40 bpm)
- Course
- Usually improves in parallel with motor & sensory function
- Rarely any long-term autonomic dysfunction
- Frequency: 60%
- Progression
- Nadir: Mean at 9 days
- General definition: Progression for < 4 weeks
- Treatment related fluctuations: More common with
- Severe disease
- Prominent sensory abnormalities
- 1% to 20% have acute onset of CIDP vs GBS: May need repeat treatment; ? Steroid responsive
- Causes of morbidity
- Weakness
- Respiratory failure
- Pneumonia or sepsis
- Dysphagia
- Thromboembolism
- Corneal exposure
- Sensory
- Pain: 2° neuropathy or immobility
- Autonomic
- Cardiac Arhythmias
- Labile blood pressure
- Hypersensitivity to cardiovascular medications
- Weakness
- Prognostic factors
- Mechanical ventilation needed
8
- Rapid disease progression
- Bulbar dysfunction
- Facial weakness: Bilateral
- Dysautonomia
- Pulmonary function testing
- Vital capacity < 20 ml/kg
- Decrease from baseline > 30%: Vital capacity or Respiratory pressure
- Residual disability greater
- Clinical prognostic factors for residual disability
- Increasing age (especially > 40 to 60)
- Weakness
- Severe at nadir
- Need for ventilatory support
- Rapid development
- Complete areflexia in the acute stage
- Diarrhea prodrome: Especially with Plasma exchange treatment
- Lack of treatment with plasma exchange or IV Ig
- Longer time to improvement
- Initial improvement > 21 days
- Disability present at 12 to 18 months
- Laboratory prognostic factors for residual disability
- Axon loss
- Low compound motor action potential (CMAP) amplitudes
-
< 20% of normal
- ? Lack of demyelinating features
- Low compound motor action potential (CMAP) amplitudes
- Serology
- ? Serum IgG vs GM1 ganglioside
- ? Preceding Campylobacter jejuni infection (past 4 weeks) or diarrhea
- Recent CMV infection
- Serum Albumin: Low
31
- Respiratory weakness: More
- Severe weakness at 1 and 6 months: More
- Inability to walk: More
- Albumin levels may be reduced after IVIg treatment
- Axon loss
- Clinical prognostic factors for residual disability
- Mechanical ventilation needed
8
- Death
- Frequency: 3% to 10%
- Causes: Pneumonia; Iatrogenic hypotension
- Associations: Mechanical ventilation; ? Autonomic dysfunction
- Associated Systemic Disorders
- Infections: Prodromes
- Viral
- Cytomegalovirus (CMV)
- Epstein-Barr virus (EBV) ± Hemophagocytic syndrome
- HIV
- Influenza
- Bacterial
- Viral
- Lymphoma
- Sarcoid
- Porphyria
- Hyponatremia
- Mildly reduced Na+ in 7% to 26%
- Severely reduced Na+ (SIADH) (105 to 120 mEq/L) may occur
- No relation to degree of severity of GBS
- Renal
- Common: Mild transient proteinuria
- Rare: Glomerulonephritis
- Cardiac
- Arhythmias: 10% to 75%
- EKG changes: > 50%
- Serum CK: High in 33%; Up to 4x normal
- Treatment
- Immunomodulation
- Plasma Exchange or IV IgG
- Definitely indicated
- Patients with: Inability to walk
- Timing: 1st 2 weeks of disease
- Decision between IV IgG & PE: Depends on individual features of patient & disease
- Probably indicated
- Milder weakness
- Early in disease course
- Doses
- Plasma Exchange: 5x over 8 to 10 days
- IVIg: 2g/kg total over 2 to 5 days
- Plasma Exchange vs IV IgG
- Often provide similar degrees of benefit
- Exception
- Associated IgG vs GM1, GM1b, or GalNAc-GD1a gangliosides: IVIg more effective
- 2nd treatment course: No additional benefit 40
- Definitely indicated
- Not Corticosteroids
- ?? Eculizumab
32
- Timing: Within 2–13 days of disease onset
- Dose: 900 mg q week for 4 weeks with antibiotic prophylaxis
- Concurent IvIg treatment
- No benefit at 4 weeks
- Possibly more improvement at later times (20 to 24 weeks) after treatment
- Plasma Exchange or IV IgG
- Ventilatory support
- Avoid anti-hypertensive medications
- ? Sub-cutaneous heparin
- No evidence of benefit with moderate or mild weakness
- ? Lower Risk of thromboembolic events in intensive care with severe weakness
- Immunomodulation
- Onset
- Childhood GBS
- Ages: Neonatal to Teens
- Onset
- Lower extremity ® Generalized weakness
- Pain & Paresthesias (60%): Lower limbs or Back
- Miller-Fisher syndrome: 1% of childhood AIDP
- CNS signs: More frequent; At onset
- Bladder dysfunction
- Mental status changes & headache
- Pain & meningismus (30%)
- Ataxia: Gait
- Occasional: Papilledema (< 5%)
- Recovery
- Often more rapid than adults
- Disability at 1 year: Rarely full recovery
- Residual disability (~30)%: Foot drop, Pes cavus, Tremor
- Nerve conduction studies in GBS: Demyelination ± Axonal loss
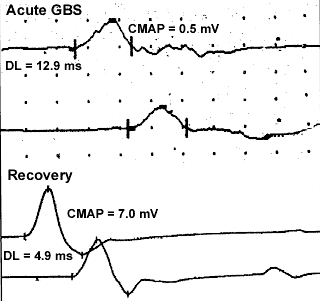
M. Al-Lozi- Common early (< 1 week from onset) features
9
- Reduced H reflex (97%)
- SNAPs: Upper extremity (61%); Sural may be preserved
- Reduced F-waves (84%)
- Other electrodiagnostic features: Demyelination
- Overall
- Features less common in 1st 5 to 7 days of disease
- Increased frequency when multiple nerves studied
- Features of demyelination for more specific diagnosis
- One abnormality in 2 different nerves
- Nerves: Median & Ulnar or Peroneal
- Specific features
- Distal motor latency: > 150% upper limit of normal
- Motor NCV: < 70% lower limit of normal (? CIDP)
- F-wave latency: > 150% upper limit of normal
- CMAP amplitude decay: > 10% to 30%
- CMAP temporal dispersion
> 300% upper limit of normal (Distal) - CMAP temporal dispersion
- > 150% upper limit of normal
- Distal: Proximal
- Overall
- Motor conduction block probably causes acute weakness
- Locations
- Proximal nerve roots
- Along course of nerve
- Distal near motor nerve terminals
- Locations
- Compound motor action potentials (CMAPs):
- Often become progressively small
- Small CMAPs may indicate
- Axonal loss or distal conduction block
- Poor prognostic sign
- EMG: Fibrillations & Positive sharp waves
- Onset: 2 to 4 weeks
- Peak: 2 to 3 months
- Common early (< 1 week from onset) features
9
- Acute immune neuropathies: Humoral Immunity
- IgG vs GM1, GM1b or GalNAc-GD1a: Motor syndromes (AMAN)
- United States: < 2%
- Japan, China, Australia & ? Europe: 10% to 20%
- IgM or IgG vs Heparan sulfate: 20%
- IgG vs other glycolipids: 10% to 30%
- IgM or IgG vs PMP-22: Probably testing artefact; Not specific for GBS
- IgM or IgG vs Tubulin: 3%
- Tumor necrosis factor-α: High serum level correlates with demyelination
- IgG or IgM vs molecules at nodes of Ranvier or paranodes (30%)
27
- Target molecules
- Gliomedin (GLDN)
: Demyelinating GBS
- Neurofascin-186
: Axonal acute immune neuropathies
- IgG1 vs Gelsolin-3: AIDP with bifacial weakness 45
- Gliomedin (GLDN)
- Target molecules
- IgG vs GM1, GM1b or GalNAc-GD1a: Motor syndromes (AMAN)
- GBS: Laboratory
- CSF: Albumino-cytological dissociation
- Protein
- Early (1st 2 days): Usually (85%) normal
- Later
- High; 66% in 1st week; 82% in 2nd week
- Highest with most slowing of NCV
- Cells: Normal (~90%)
- Usual (90%): Normal (< 10 cells/mm3)
- May be high with associated disorder present
- Oligoclonal bands: 10% to 30%
- Protein
- Hematology: Only abnormal with associated infection or other disorder
- Serum CK: Higher in patients with pain
- ESR: Usually < 50 mm/hr
- Mild proteinuria: 25%
- Liver function test: Abnormal in 10%
- WBC: Most commonly normal; > 20,000 only with associated infections
- HLA types: Class II associations with AIDP in northern Chinese patients
14
- General
- Regions important in peptide binding and T cell recognition
- Associated with other diseases with pathoimmunological basis
- No class II associations found for AMAN
- Susceptibility: DQβRLD55–57/ED70–71 & DRβE9V11H13
- Protection: DQβRPD55–57 epitope
- General
- CSF: Albumino-cytological dissociation
- Other: Books by GBS patients or relatives (PDF file)
 Putnam 1909 |
Acute Motor (Axonal) Neuropathy (AMAN)
- Epidemiology
- Geography
- More common: Japan, China, Mexico & 3rd world countries
- Rare: Most parts of United States
- Occasional: Europe
- Onset age: More common in children
- Male = Female
- Seasonal peaks
- Mexico & China: July to September (Rainy season)
- Japan: March for C jejuni + patients
- Geography
- Prodrome
- Gastrointestinal: Diarrhea
- + Campylobacter jejuni titers in 67%
- Campylobacter jejuni in GBS
- Serotype associations
- O-19 (HS:19) type
- > 50% when Campylobacter culture positive
- Risk of acute neuropathy: 1 in 158
- 6x greater than after other Campylobacter infections
- Common in China & Japan
- Different isolates clonally related
- Commonly bind cholera toxin
- O-41 (HS:41): Over represented in South Africa & Mexico
- Carbohydrate epitope associations: GD1a but not clearly GM1
- O-19 (HS:19) type
- Higher frequency of TNF-α
polymorphism
- Campylobacter jejuni cst-II polymorphisms: Neuropathic strains
18
- cst-II: Gene encoding sialyltransferase

- cst-II (Thr51)
- More common than enteritic strains
- Express GM1 (92%) & GD1a (91%) epitopes
- Associated with related antibodies & Limb weakness
- cst-II (Asn51)
AMAN: After recovery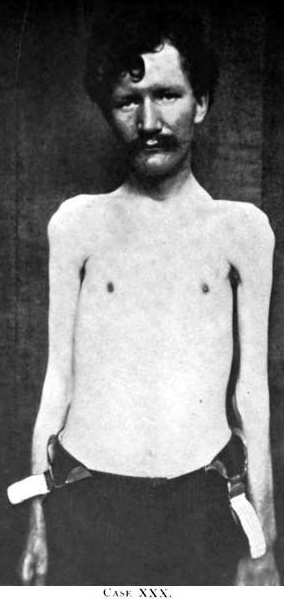
Putnam 1909
- 83% express GQ1b epitope
- Associated with
- Anti-GQ1b antibodies
- Bulbar palsy
- Ophthalmoparesis
- cst-II: Gene encoding sialyltransferase
- Serotype associations
- Upper respiratory
- Haemophilus influenzae infection
5
- Geography: Japan
- Frequency: 13%
- Organism type: Non-typable; Contains GM1-like structure
- Infection preceded disease by 4 to 12 days
- Serum antibodies: IgG vs GM1 & GD1b ganglioside common (83%)
- + CMV titers in 0%
- Haemophilus influenzae infection
5
- Zika virus
- ? Treatment with parenteral ganglioside mixture
- Acute axonal motor syndromes reported
- Onset 5 to 15 days after Rx
- Associated serum IgG anti-GM1 antibodies
- Onset 5 to 15 days after Rx
- Acute axonal motor syndromes reported
- Gastrointestinal: Diarrhea
- Clinical
20
- Weakness
- Distal: More severe than proximal
- Finger extensors: May be selectively weak
- Arms > Legs
- Proximal: Mild weakness common (85%)
- Symmetric: Usually
- Cranial Nerves (6% to 25%)
- Facial weakness: Occurs but less commonly than in AIDP
- Respiratory failure
- Uncommon
- Less than in classic demyelinating GBS
- Distal: More severe than proximal
- Sensory
- Reflexes
- Usual: Reduced in proportion to strength
- Occasional: Preserved; Hyperreflexia in some
26: Associated with
- Milder disease
- Serum IgG vs GM1, GM1b, GD1a or GalNAc-GD1a
- Progression
17
- Nadir: Mean at 6 to 12 days; May be as short as 2 days
- Shorter course than demyelinating GBS
- Treatment
- IV Ig: Anecdotal evidence
- Plasma Exchange: May be effective with Reversible Conduction Failure 43
- Not Corticosteroids
- Recovery
- Significant improvement in strength over 1 to 2 months
- ? More rapid with
- Haemophilus influenzae prodrome
- Reversible Conduction Failure 43
- Weakness
- Laboratory
- Nerve conduction studies
- Axon loss (CMAP amplitude ↓) >> Demyelination
- May have: Prolonged distal latencies or Conduction block
- Occurs: Early; 1st 2 weeks
- Disappears: At later times
- Less common than in AIDP
- Reversible Conduction Failure (RCF)
42
- Features
- Rapid recovery of: Conduction velocity & CMAP amplitudes
- No evidence of temporal dispersion
- More common with AMAN than AIDP
- May suggest: More favorable prognosis; Less severe at 1 month
- AIDP + RCF: Younger; Clinically more like AMAN
- Features
- Serum Antibodies
25
- IgG vs GM1 ganglioside (40% to 50%)
- IgG reactivity to both GM1 & GM1b gangliosides
- More strongly associated syndromes
- Distal motor
- Rapidly progressive
- Treatment response: IVIg, not plasma exchange
- More strongly associated syndromes
- IgG subclass: Associated with prodrome & speed of recovery phase
16
- IgG1
- Preceding gastroenteritis
- Campylobacter jejuni serology +
- Slow recovery (> 1 month)
- IgG3
- Preceding respiratory infection
- Rapid recovery within 1 month
- IgG1
- Epidemiology
- Common: Japan, China & 3rd world countries
- Rare: Most GBS-like sydromes (AIDP) in US & Canada
- Pathophysiology of IgG anti-GM1 antibodies
19
- May bind to: Nodes of Ranvier & Paranode of axons
- Induce binding of C3 complement
- Nodes of Ranvier: Disrupts Na+ channel clusters
- Paranode
- Detachment of paranodal myelin terminal loops
- Caspr: Absent or reduced
- ? Anti-GM1 & other antibodies block nerve conduction
- IgG reactivity to both GM1 & GM1b gangliosides
- IgG vs GalNAc-GD1a: Similar to syndrome with IgG vs GM1 ganglioside
- Campylobacter jejuni infection
- Acute motor neuropathies
- Distal predominant weakness & Sparing of the cranial nerves
- Rapidly progressive, severe weakness
- Poor recovery
- Other antibodies in AMAN syndromes
- IgM vs GM1 ganglioside (30%)
- IgG vs GD1a ganglioside (12% to 60%)
- May be 2° to IgG binding to GalNAc-GD1a contaminant in GD1a
- May be associated with AMAN or AIDP
- IgG vs GM1b (15%)
- IgM vs GalNAc-GD1a ganglioside
- Antibody associations: Other
- More common with prodromal infections
- Campylobacter jejuni
- Haemophilus influenzae
- Time course: Highest titers at disease onset; Fall over weeks 15
- Geography: Uncommon in many areas of US
- More common with prodromal infections
- IgG vs GM1 ganglioside (40% to 50%)
- MRI: Ventral root enhancement
Acute Motor Neuropathy 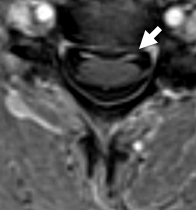
From: R Bucelli
MRI: Ventral root
enhancement (Arrow)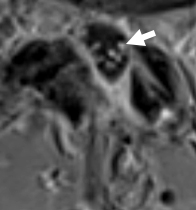
From: R Bucelli
MRI: Cauda equina
enhancement (Arrow) - Pathology
- Motor nerve terminal degeneration
- Severe weakness: More proximal axonal damage
- Ventral root: Axon loss
- Macrophage invasion of motor nodal axolemma: Focal axon damage
- Sensory nerves normal
- Nerve conduction studies
- Variant syndrome: Acute Motor-Sensory Axonal Neuropathy (AMSAN)
Guillain-Barré-like syndrome with serum IgM binding to GalNAc-GD1a ganglioside 2
- Epidemiology
- Japanese patients
- Younger onset: Mean 3rd decade
- ? Female > Male
- Clinical
- Prodrome
- GI: Campylobacter jejuni (75%)
- Respiratory: Cytomegalovirus
- Weakness: Mild; Proximal + Distal
- Sensory: Paresthesias ± Loss (75%); More common with CMV prodrome
- Cranial nerves: Facial weakness (27% to 80%); More common with CMV prodrome
- Recovery: Good at 1 month; Better than with IgG or no antibodies
- Prodrome
- Electrodiagnostic
- Demyelination in many (38% to 64%)
- Axonal loss uncommon
- Serum antibody
- IgM vs GalNAc-GD1a ganglioside
- Cross reactive with GM2 ganglioside: 100% with CMV prodrome; 50% with GI prodrome
- Patients with GI prodrome commonly have other IgM antibodies as well: anti-GM1 & GD1b
- Also see
- Pathology
Miller Fisher Syndrome 6
|
|
(Sub)Acute Sensory Neuropathies 24
|
Autonomic Acute ataxic: GD1b antibodies; Acute or Relapsing Paracarcinomatous (anti-Hu antibodies) Polyradiculopathy (AISP) Sensory Guillain-Barré Sjögren's (SSA or SSB antibodies) Small fiber neuropathy/neuronopathy, Acute Toxic Cis-platinum Pyridoxine intoxication Also see: Acute neuropathies |
Acute Neuropathy: Sensory Polyneuropathy with some Demyelinating features (Sensory Guillain-Barré)
- Clinical
- Antecedent illness: Flu or URI in some
- Onset
- Over Days to Weeks
- Paresthesias & Pain
- Sensory
- Loss
- Pansensory
- Proximal + Distal
- Paresthesias & Pain
- Loss
- Autonomic dysfunction: Mild
- Motor: Normal; Occasional mild weakness
- Tendon reflexes
- Usually absent
- Rare patients may have preserved tendon reflexes
- ? Central branch of sensory neuron damaged rather than cell body
- ? Preserved Ia afferents to motor neurons
- Course
- Monophasic
- Prognosis: Often incomplete recovery
- Laboratory
- Nerve conduction studies
- Sensory potential amplitudes: Usually absent; May be reduced or normal
- Conduction velocities: Relatively normal
- Demyelinating features: Prolonged distal latencies in some patients
- CSF: Protein high; No cells
- Pathology
- Nerve biopsy: Axonal loss; Mild inflammation
- Autopsy: Lymphocytes in nerves & Dorsal roots
- Epstein-Barr virus titers elevated in some patients 1
- Nerve conduction studies
Acute neuropathy: Bickerstaff brainstem encephalitis 13
- Epidemiology: Most reports from Japan
- Antecedent illness (92%): Most commonly upper respiratory infection
- Age: 3 to 91 years
- Onset: Diplopia or gait disorder most common
- Clinical: Brainstem signs
- Reduced consciousness (74%): Drowsy, stupor or coma
- Ataxia: Often trunk & limb
- Eyes
- Ophthalmoplegia, external (100%): Relatively symmetric
- Pupil disorders (34%)
- Ptosis (29%)
- Nystagmus (27%)
- Other cranial nerves
- Facial diplegia (45%)
- Bulbar weakness (34%)
- Weakness: Flaccid tetraparesis (60%); Respiratory failure
- Pyramidal signs
- Tendon reflexes: Variable; Hyperreflexia to Absent
- Plantar responses (40%): Extensor
- Sensory loss
- Small fiber (31%)
- Large fiber (16%)
- Hemisensory loss
- Course
- Monophasic
- Often good prognosis: Complete remission in 51%; Death 4%
- Laboratory
- Serum IgG binding to GQ1b ganglioside (66%)
- Electrophysiology
- Motor axon degeneration
- Central involvement
- MRI: CNS abnormalities
- High intensity T2 signal
- Locations: Brainstem, Thalamus, Cerebellum
- CSF
- Cells: Usually normal; Occasionally high WBCs (37%)
- Protein: High in 59%; Higher in 2nd week than 1st
- Pathology
- Perivascular lymphocytic infitration
- Edema
- Glial nodules
Acute neuropathy: Pharyngo-Cervical-Brachial Variant 21
- Prodromal associations: Within 4 weeks of illness
- Upper respiratory infection (70%)
- Diarrhea or other GI (30%)
- Infection: Campylobacter jejuni > Cytomegalovirus
- Clinical
- Bulbar palsy
- Dysphagia
- Weakness
- Neck
- Arms: Proximal; Often initial symptom
- Legs: Relatively preserved; Hip flexors most involved
- Face: 50%
- Sensation: Reduced or Normal
- Tendon reflexes: Reduced, especially in arms
- Cranial nerves
- External ophthalmoplegia (50%)
- Ataxia (40%)
- Autonomic(20%): Especially heart rate & bladder dysfunction
- Progression: 1 to 3 weeks
- Overlap syndromes
- GBS: 48%
- Miller-Fisher (MFS)-like: 30%
- None: Pure PCB in 13%
- Bulbar palsy
- Laboratory
- CSF protein: High in some (42%)
- NCV: Abnormal motor conduction or late responses
- Antibody
- IgG binding to GT1a or GM1b gangliosides without binding to GQ1b (65%)
- IgG binding to GQ1b: Common with MFS overlap
Acute neuropathy: Bulbar Palsy Variant 29
- Epidemiology: Asian patients
- Clinical
- Onset age: 18 to 54 years
- Bulbar: Weakness
- Gait: Ataxia (82%)
- Face palsy (55%)
- Ophthalmoplegia (82%): Abducens or Complete
- Strength: Normal
- Tendon reflexes: Absent (90%)
- Paresthesias (55%): Limbs
- Dizziness (90%)
- Course: Monophasic over weeks to 3 months
- Laboratory
Acute Immune Neuropathies: Facial Diplegia Variant (BFP) 22
- Epidemiology
- Japanese & North American patients
- Prodrome
- Infectious symptoms in previous 4 weeks
- Frequency: 86%
- URI or Fever most common
- Infectious symptoms in previous 4 weeks
- Clinical
- Onset
- Age: 18 to 65 years
- Limb numbness
- Bifacial weakness
- Progressive: Over weeks
- Asymmetric: Often
- Paresthesias
- Most patients
- Distal limbs
- May persist for months
- Strength: Normal or Mildly reduced
- Tendon reflexes: Reduced or Absent
- Other cranial nerves: Often normal
- Course
- Monophasic
- Nadir at 4 weeks
- Improvement over months
- Differential diagnosis
- Onset
- Laboratory
- CSF: Albumino-cytologic dissociation
- Protein: 51 to 256
- Cells: 1 to 8
- Nerve conduction studies
- Demyelination (64%)
- Evolves with disease course
- Proximal or Distal
- Distal latencies & F-waves: Prolonged
- Recovery toward normal over months
- SNAPs: Amplitude Moderately reduced
- Demyelination (64%)
- Infection-related
- CMV antibodies: 35%
- Other possible infection: B. burgdorferi
- Auto-Antibodies
- CSF: Albumino-cytologic dissociation
Acute Neuropathy: Sensory Polyradiculopathy 37
- Epidemiology: 1 patient
- Clinical
- Onset age: 19 years
- Prodrome: Headache & Fever
- Sensory loss
- Arms & Legs
- Distal to Upper arms & Thighs
- Vibration & Proprioception
- Ataxia: Limb & Gait
- Areflexia
- Motor & Cranial nerves: Normal
- Course
- Progression: Over 3 weeks
- Recovery: Over 6 months
- Laboratory
- NCS & EMG: Normal
- SEP: Abnormal cervical & cortical respponses
- CSF: Protein 94; 1 cell
- Brain & spine MRI: Normal
- Serum antibody: IgG vs Contactin-1
Return to Neuromuscular Syndromes
Return to Acute Neuromuscular disorders
Return to Neuromuscular Home Page
References
1. Muscle Nerve 1999;22:1607-1610
2. Brain 2000;123:116-124. J Neuroimmunol 2001;113:260-267
3. Neurology 2000;54:1000
4. Neurology 2000;54:1661-1665
5. Brain 2000;123:2171-2178
6. Neurology 2001;56:1104-1106, Front Neurol 2023;14:1250774
7. Acta Neurol Scand 2001;103:259-260, J Integr Neurosci 2022;21:81
8. Arch Neurol 2001;58:893-898
9. Arch Neurol 2001;58:913-917
10. Neurology 2001;57:736-738
11. Neurology 2001;57:686-691
12. Neurology 2001;57:1755
13. Muscle Nerve 2002;26:845-849, Brain 2003;126: Online July
14. J Immunol 2003;170:3074–3080
15. J Neurol Sci 2003; Online April
16. Neurology 2003;60:1514–1518
17. Neurology 2003;61:471–474
18. Neurology 2005;65:1376–1381
19. J Peripheral Nervous System 2007;12:238–249
20. Neurol India 2009;57:282-286
21. J Neurol Neurosurg Psychiatry, 1999;66:513-516, J Neuroimmunol, 2000;105:195-201, J Neurol Neurosurg Psychiatry 2002;72:767–771, J Neurol Sci 2004; Online March
22. J Neurol 2009 Jul 25, Muscle Nerve 2016; Online January, J Peripher Nerv Syst 2026;31:e70106
23. Neurology 2010;75 Online Sept
24. Muscle Nerve 2011; September
25. J Neurol Neurosurg Psychiatry 2011; Online October
26. J Neurol 2011; Online Dec
27. J Peripher Nerv Syst 2012;17:62-71
28. Vaccine 2015 Mar 3
29. Neurology 2015 Dec 30
30. Euro Surveill 2014;19(9)
31. JAMA Neurol 2016 Dec 27
32. Lancet Neurol 2018 Apr 20
33. Neurol Neuroimmunol Neuroinflamm 2020;7:e833, PLoS Negl Trop Dis. 2020 Dec 17;14(12):e0008032, J Peripher Nerv Syst 2021 Jul 12
34. Brain 2020 Dec 14
35. Neurology 2020 Dec 14
36. Ann Neurol 2020 Dec 9
37. Muscle Nerve 2020 Dec 21
38. J Neuroimmunol 2021 Jul 1
39. Muscle Nerve 2021 Sep 10
40. J Neurol Sci 2022 Jul 29
41. Cureus 2023;15:e43111
42. Sci Rep 2022;12:18562
43. J Clin Neuromuscul Dis 2019;21:35-41
44. J Peripher Nerv Syst 2025;30:e12683
45. Ann Neurol 2026 Mar 24
5/19/2026