Home, Search, Index, Links, Pathology, Molecules, Syndromes,
Muscle, NMJ, Nerve, Spinal, Ataxia, Antibody & Biopsy, Patient Info
|
Home, Search, Index, Links, Pathology, Molecules, Syndromes, Muscle, NMJ, Nerve, Spinal, Ataxia, Antibody & Biopsy, Patient Info |
![]() Recent evidence shows that several
pure motor
neuropathy syndromes can
be distinguished from amyotrophic lateral sclerosis (ALS). Identification of these motor
neuropathy syndromes is important as, in contrast to ALS, they are often immune-
mediated and treatable.
The motor neuropathy syndromes usually have characteristic, but not unique, patterns of weakness and
no upper motor neuron signs. Additional
laboratory evaluation, including electrodiagnostic studies and measurement of serum
autoantibodies, is usually needed to clearly distinguish these disorders from other
demyelinating neuropathies and motor neuron syndromes.
Recent evidence shows that several
pure motor
neuropathy syndromes can
be distinguished from amyotrophic lateral sclerosis (ALS). Identification of these motor
neuropathy syndromes is important as, in contrast to ALS, they are often immune-
mediated and treatable.
The motor neuropathy syndromes usually have characteristic, but not unique, patterns of weakness and
no upper motor neuron signs. Additional
laboratory evaluation, including electrodiagnostic studies and measurement of serum
autoantibodies, is usually needed to clearly distinguish these disorders from other
demyelinating neuropathies and motor neuron syndromes.
Historical Aspects of Lower Motor Neuron Syndromes
The original descriptions of pure motor syndromes without upper motor neuron
signs were probably cases of "progressive muscular atrophy" in the writings of
Duchenne, Aran and others during the 19th century . Benign, focal motor neuron
disorders, such as monomelic amyotrophy,
were subsequently reported . These
syndromes were usually considered as variants of ALS, as early pathological studies
suggested that the primary focus of the disease was on cell bodies in the ventral horn.
A pathological report by Rowland et al. first documented that a patient with a pure motor syndrome could have the primary site of disease along the course of the axon. This patient, with a lower motor neuron (LMN) syndrome and a serum IgM M-protein, had damage to motor axons but not cell bodies. Motor neuropathies were first diagnosed during life by electrodiagnostic testing. Nerve conduction studies showed blockade of impulses at focal sites along the course of motor axons (motor conduction block) providing strong evidence that the primary site of disease lay in the peripheral nerve rather than the cell body. The phenomenon of conduction block had been described earlier in patients with sensory-motor neuropathies (chronic inflammatory demyelinating polyneuropathy (CIDP)) . Conduction block was thought to result from focal regions of immune-mediated demyelination along the course of the nerve.
In 1986 a patient was reported with a LMN syndrome without conduction block, but with a
serum IgM M-protein that bound to GM1 ganglioside. In this instance the association of
the motor syndrome with an autoantibody directed against a neural antigen suggested
that the disorder might be immune-mediated. However, attempts at
immunosuppression had no effect on the progressive disease in that patient. A clinical
response to immunotherapy remains a "gold standard", without which it is difficult to
argue that a syndrome is immune mediated. In 1988 two patients with a multifocal
motor neuropathy, motor conduction block, and serum IgM anti-GM1 antibodies were
reported to improve after treatment with cyclophosphamide . It now appears that either
motor conduction block or serum anti-GM1 antibodies alone can be markers for
patients with LMN syndromes that often improve after immunomodulating therapy.
Multifocal motor neuropathy (MMN) and motor syndromes with serum anti-GM1
antibodies.
A. Clinical Syndromes
The immune-mediated motor neuropathies are characterized by
asymmetric,
slowly progressive weakness that most commonly
begins in the arms. The age of
onset is generally between 20 and 75. Men are affected somewhat more commonly
than women. Motor findings include asymmetric weakness and variable degrees of
atrophy. Patients with prominent
conduction block may present with weakness in
muscles with relatively normal bulk. Rarely, patients have had cranial nerve signs
including external ophthalmoplegia and unilateral tongue weakness and atrophy.
Some patients report paresthesias, but sensory signs are usually absent or clinically
insignificant. In regions with normal strength
tendon reflexes are often preserved. In
areas of weakness, reflexes may initially be normal but can become reduced with
progression of the disease.
Fasciculations are not uncommon, and may
add to diagnostic confusion between MMN and variants of
amyotrophic lateral sclerosis (ALS)
with only lower motor neuron signs. However, hyperreflexia and spasticity typical of
ALS never occur in MMN.
|
|
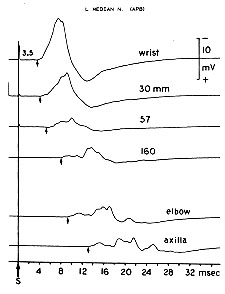
Conduction block Amount of block increases with more proximal stimulation. |
Galβ1-3GalNAcβ1-4Galβ1-4Glcβ1-1'Ceramide
3
|
Neu5Acα2
|
A major issue regarding clinical testing for anti-GM1 antibodies is differences in the methodology used in their measurement. Technical validation of methodology by identifying serums with high antibody titers is not sufficient. Laboratories must also document the sensitivity and specificity of anti-GM1 antibody testing methods by clinical correlation studies using serums from patients with motor syndromes.
In our laboratory the sensitivity of antibody (IgM vs Co-GM1 and NP-9)
testing for MMN is now 85% to 90%. In general, high titers of serum
IgM anti-GM1 antibodies should be detected in:
Improvement in strength after treatment with
HIG (for example 1 g/kg/day x 2 days) is common (50% to 70% of cases),
but the length of benefit is variable, lasting from 2 weeks to 6 or more months.
The dose and frequency of subsequent treatments is based on individual patient
response.
Motor neuropathies and motor neuron disorders. Some motor
neuropathies have been classified as ALS variants, with predominantly LMN signs and
axonal changes on electrodiagnostic studies. Certain features can aid in the
differentiation between motor neuropathies and ALS. Patients with motor
neuropathies may have preserved reflexes in weak muscles, but overt spasticity and
bulbar features are conspicuously lacking. This is in contrast to patients with ALS who
often have prominent upper motor neuron and bulbar findings. The prolonged course
that is often noted in patients with motor neuropathies also helps to differentiate their
syndromes from typical ALS. Acquired motor neuropathies most often produce
asymmetric weakness. This pattern is usually clinically distinct from the proximal
symmetric weakness that characterizes most of the hereditary spinal muscular
atrophies.
Several lower motor neuron syndromes
have been described that are of
uncertain etiology and could be disorders of the motor axon or cell body. A majority of
patients with D-LMN syndromes have neither evidence of peripheral nerve
demyelination nor serum anti-ganglioside antibodies. These patients tend to have
more rapidly progressive weakness than is typical for the immune-mediated motor
neuropathies. In contrast to typical ALS, many D-LMN patients never develop bulbar
dysfunction. There are no reports of response to immunosuppressive treatment in
D-LMN patients with neither demyelination nor serum autoantibodies. Some
patients develop progressive asymmetric lower motor syndromes with predominant
early weakness in proximal musculature (P-LMN syndromes). Characteristic features
include late-age onset, male predominance (85%) and initial signs of weakness in the
upper extremities (80%). Progression is slow. Weakness is often confined to one or
two extremities for 3 to 5 years. Electrodiagnostic studies show only evidence of
axonal loss. Some patients with P-LMN syndromes (30%) have selective serum
antibody binding to GA1 ganglioside. However, there is no evidence that P-LMN
syndromes respond to immunosuppressive treatment.
Monomelic amyotrophy,
a syndrome that affects mainly young (15 to 25 years)
males (80%), presents with weakness of the distal musculature of one upper extremity
that progresses for 1 to 2 years and then remains stable. Occasional patients develop
weakness in the opposite limb, mild sensory symptoms or tremor. Electrodiagnostic
studies show denervation in the affected limb. There are no associated serum
antibodies.
Rare patients with paraneoplastic
LMN syndromes have been reported. The best described of these is a
subacute motor neuronopathy associated
with lymphomas, such as Hodgkin’s disease.
Progressive, asymmetric weakness develops, most
severely in the legs, at times when the neoplasm is in remission or
during irradiation. The weakness is rarely severe, and often stabilizes
or improves over a period of months to years.
Pathological studies show a loss of motor neurons in the ventral horn
of the spinal cord and some involvement of sensory tracts. LMN involvement has also
been described as an occasional part of the paraneoplastic encephalomyelitis and
sensory ganglionopathy syndromes that occur in association with anti-Hu antibodies.
There is no clear evidence that there is an increased incidence of paraneoplastic
"typical" ALS syndromes, with upper and lower motor neuron involvement. A few
patients with ALS-like syndromes and neoplasms, including renal cell, lung and
lymphoma, have been reported to improve or stabilize after treatment of the cancer.
Other immune-mediated demyelinating neuropathies have more sensory
involvement and are rarely confused with MMN.
Anti-MAG neuropathies are
often associated with weakness, but sensory loss is most often the presenting, and
disabling, feature of the disease. Neuropathies with
anti-sulfatide antibodies and
GALOP syndrome
have even more predominant sensory involvement.
POEMS
syndrome may produce severe weakness but this is accompanied by prominent
sensory loss and systemic signs.
During the next 10 months there was progressive weakness and loss of reflexes
despite high dose prednisone therapy and 11 treatments with plasma exchange. Six
monthly treatments with two plasma exchanges followed by intravenous
cyclophosphamide (1 gm/M2 ) were followed by improvement, beginning after 3 to 4
months, and progressing to nearly normal strength over the next year. She remained
stable, off all medications, for 3 years. She then noted mild recurrent weakness in the
right hand in a distribution similar to that at disease onset 5 years before.
COMMENT: Asymmetric weakness, developing distally in an arm or hand, is the
most common pattern of early involvement in MMN. Reflexes are often normal early in
the disease course. Significant sensory signs are rare, but patients occasionally note
symptoms such as paraesthesias, or even abnormal taste sensations. Prednisone
treatment is rarely effective, and is often associated with rapid exacerbations of
weakness. The decision to use cyclophosphamide was only made when 1. it became
clear that the patient had developed significant disability, and 2. there were clear signs
that an immune disorder was present, including conduction block and high serum titers
of IgM anti-GM1 antibodies. Improvement in strength after cyclophosphamide begins
late, often 3 to 6 months after beginning therapy, and continues for up to a year after
the end of the treatment course.
CASE 2: A 52 year old male noted a right foot drop. During the next 6 months
weakness and cramps became progressively worse in the right leg and also developed
in the left leg. On examination there was asymmetric weakness, predominantly in the
legs. Muscle tone was normal. Cranial nerves were normal. Tendon reflexes were
absent at the right ankle, but 2+ elsewhere. Sensation was normal. Electrodiagnostic
testing showed denervation in thoracic paraspinous muscles and both lower
extremities. Nerve conduction velocities were normal. No serum anti-GM1 antibodies
were detected.
COMMENT: This patient has relatively rapidly progressive weakness with
evidence of involvement of lower, but not upper, motor neurons. The clinical course is
more rapid than that usually seen in MMN. The most appropriate diagnostic categories
would be the older term, “progressive muscular atrophy”, or, descriptively, “lower motor
neuron syndrome”. Such patients may not develop bulbar, or upper motor neuron,
signs and never meet diagnostic criteria for ALS. Denervation in the thoracic
paraspinous muscles is more suggestive of a motor neuron disease than a motor
neuropathy. With no evidence of demyelination or serum antibodies to suggest an
immune etiology for the syndrome, it is likely that this patient will have continued
progression of weakness that does not respond to immunosuppressive treatments.
CASE 3: A 55 year old man was referred for possible immunosuppression to treat a
lower motor neuron syndrome with anti-GM1 antibodies. He had noted increasing
difficulty climbing stairs and arising from a chair for 10 to 15 years. In recent years his
speech had become slurred and he needed more time to eat. On general examination
there was mild gynecomastia. Neurological testing showed weakness and
fasciculations of the tongue and face. The tongue showed severe atrophy. Moderate
symmetric, proximal weakness was present. Tendon reflexes were difficult to elicit.
Sensation was reduced to all modalities distally in the feet. Electrodiagnostic testing
showed chronic denervation, most prominent in the face, tongue, and proximal
muscles. Repeat anti-GM1 antibody testing showed a pattern of polyreactive serum
IgM binding to GM1 ganglioside and to histone H3 at titers of about 1,500.
COMMENT: The patients weakness was proximal and symmetric, more typical of
inherited motor neuron disorders than of acquired motor neuropathies. Although the
patient had high titers of anti-GM1 antibodies, the pattern of binding was polyreactive.
Polyreactive antibodies are not specific for immune motor syndromes, and are also
found in 3% to 5% patients with ALS and adult-onset spinal muscular atrophies.
Further testing revealed an excessive number of trinucleotide repeats in the androgen
receptor, a finding consistent with X-linked hereditary bulbo-spinal muscular atrophy.
This case emphasizes that determination of the specificity of anti-GM1 antibodies helps
to determine their clinical relevance. The polyreactive antibodies in a patient with
features atypical of a motor neuropathy were not, of themselves, an indication for
immunosuppressive therapy.
Lopate G, Pestronk A. Chronic immune demyelinating neuropathies. Seminars in
Neurology 1994;14:131-136.
Parry G. Motor neuropathy with multifocal conduction block. In: Dyck PJ, Thomas PK,
Griffin JW, Low PA, Poduslo JF, eds. Peripheral Neuropathy. 3rd ed. Philadelphia:
W.B. Saunders Company 1993 pp 1518-1524.
Pestronk, A., Choksi, R. Multifocal motor neuropathy: Serum IgM anti-GM1
ganglioside antibodies in most patients detected using covalent linkage of GM1 to
ELISA plates. Neurology 1997;49:1289-1292.
Pestronk A, Chaudhry V, Feldman EL, et al. Lower motor neuron syndromes defined by
patterns of weakness, nerve conduction abnormalities, and high titers of antiglycolipid
antibodies. Ann Neurol 1990;27:316-326.
Takigawa T, Yasuda H, Kikkawa R, Shigeta Y, Saida T, Kitasato H. Antibodies against
GM1 ganglioside affect K+ and Na+ currents in isolated rat myelinated nerve fibers. Ann
Neurol 1995;37:436-442.
1. at least 80% of patients with multifocal motor neuropathy, and
2. less than 1% of patients with typical amyotrophic lateral sclerosis.
A common misleading practice is citation, in test reports,
of statistics from the literature without clinical validation of the specific methods used in
a laboratory. Laboratories that cannot provide correlation data, relating patient
syndromes to results from their specific methodology, are not qualified to perform
measurements of serum anti-GM1 antibodies as a clinical test.
D. Pathogenic Mechanisms
Although the exact role of anti-GM1 antibodies in producing disease has not
been determined, there is growing evidence that they can be pathogenic. Immunization
of rabbits with GM1 may result in the production of a neuropathy with similarities to
MMN. Conduction block has been induced experimentally by the intraneural injection of
serum immunoglobulin from patients with motor neuropathy and anti-GM1 antibodies.
Anti-GM1 antibodies can alter K+ and Na+ currents in myelinated axons. Specific
anatomical patterns of binding of some anti-GM1 antibodies to peripheral nerve, spinal
cord, and motor neurons are consistent with clinical and electrophysiologic findings in
MMN. Clinically, titers of anti-GM1 antibodies decline before improvement in strength
after cyclophosphamide treatment. Finally, there is more GM1 ganglioside in myelin
from motor nerves than from sensory nerves. This could render motor fibers more
susceptible to attack by anti-GM1 autoantibodies and explain their selective
involvement in MMN and D-LMN syndromes.E. Treatment
Treatment with cyclophosphamide or human immune globulin (HIG) can produce useful functional improvement in patients with MMN.
1. The period of maximum improvement after HIG treatment should be monitored.
Subsequent treatments should be given just before a relapse is expected.
The minimum effective dose of HIG can be determined by sequentially reducing
the subsequent HIG doses by 10% until a level is found that produces somewhat
less benefit (length of time, or degree, of improved strength).
The minimum dose that produced an optimal improvement is then used for
long-term therapy.
2. Lack of improved strength after one, or at most two, treatments
(total 2 to 3 g/kg each) should be considered a treatment failure.
No further HIG should be used.
Although the side effects of HIG are usually benign, its great expense mandates
objective documentation of any benefit, including quantitative muscle
testing and functional assessment, to justify continued use.
Further studies are required to document
whether HIG treats the underlying pathogenic process in MMN or produces
symptomatic benefit while allowing the underlying immune process to progress.
Corticosteroid (Prednisone or Solumedrol)
treatment is rarely helpful in MMN and may often exacerbate weakness.
Cyclophosphamide is the only immunosuppressive medication that has been
reported to induce long-term benefit in many patients (50% to 80%) with MMN.
Unfortunately, its toxicity, especially the increased risk of neoplasia with high
cumulative life-time doses (>75 g), requires a careful analysis of the risk:benefit ratio in
each patient. Therapeutic regimens should utilize doses of cyclophosphamide that are
high enough to reduce anti-GM1 antibody titers by 60%, or more. We originally used
an initial dose of 3 g/M2 over 8 days followed by chronic oral medication (100-150 -
mg/day for 6 to 12 months). More recent experience suggests that 6 monthly
treatments with intravenous cyclophosphamide (1g/M2), each preceded by two plasma
exchanges, is equally effective, has fewer adverse effects and utilizes a 50% to 70%
lower cumulative dose of drug. This regimen produces a sustained reduction in titers of
serum anti-GM1 antibodies in approximately 60% to 80% of patients. Most patients in
whom antibody titers are reduced show functional benefit. Remission usually persists
for 1-3 years; after which, antibody titers often rise and weakness recurs. Retreatment
may then be necessary.
Deciding whether to treat patients with D-LMN syndromes without electrodiagnostic evidence of demyelination may be difficult. High titers of serum IgM anti-GM1 antibodies are a useful
indicator that a D-LMN syndrome may be immune-
mediated and treatable. Evidence of demyelination on sural nerve biopsy may also be
helpful in this regard. Measurable improvement in strength after treatment with HIG
may provide support for further immunotherapy, with agents such as cyclophosphamide
or further periodic HIG infusions.
2. Differential Diagnosis of Motor Neuropathies
The differential diagnosis of motor neuropathies includes motor neuron
disorders, hereditary and acquired, on one hand, and demyelinating
neuropathies, on the other.
Immune demyelinating Neuropathies. Although
MMN and
CIDP are both
demyelinating neuropathies, the differences in their clinical, electrophysiological and
immunologic features are more prominent than their similarities. MMN
commonly presents with distal asymmetric weakness while in CIDP, proximal symmetric
weakness is a more common finding. The remitting and relapsing course that may
occur in CIDP is uncommon in the motor neuropathies. Patients with MMN rarely have
significant sensory symptoms while in CIDP, sensory signs are the rule.
Electrophysiological testing may show conduction block in both conditions. However,
other features of demyelination such as prolonged distal latencies and slowed
conduction velocities are more prominent in CIDP. Abnormalities in sensory nerve
conduction studies are usually seen in CIDP, but not in MMN, unless complicated by
another disease process. The spinal fluid examination shows markedly increased
protein concentration in the majority of cases of CIDP while this change is rare in
patients with MMN. High titer anti-GM1 antibodies as well as more specific patterns of
autoantibody reactivity (see above) are common in MMN. In CIDP anti-GM1 antibodies
are unusual. Serum autoantibody binding to tubulin is more common. Finally,
differences in the frequency of therapeutic response to prednisone and plasma
exchange (common in CIDP, but rare in motor neuropathies) define a practical
difference in the management of the two disorders.3. Illustrative cases
CASE 1: A 42 year old woman with progressive weakness in the right hand for
one year was referred for possible motor neuron disease. During the next year the
hand weakness became more severe and also appeared in new areas of the body, first
the left hand and then the left foot. Physical examination revealed asymmetric
weakness that was predominantly distal. The thenar eminence in the left hand was
severely weak but showed no wasting. Tendon reflexes were 1+ throughout. Sensory
testing showed only a minimal reduction in vibratory sensation at the toes.
Electrodiagnostic studies showed motor conduction block in the median nerves
bilaterally between the elbow and wrist. Nerve conduction velocities were normal.
Sensory nerves were normal. Serum IgM anti-GM1 antibodies were present in high
titer (3,900; normal <600).Bibliography
Kornberg AJ, Pestronk A. Chronic motor neuropathies: Diagnosis, therapy, and
pathogenesis. Ann Neurol 1995; 37(S1):S43-S50.
Descriptions of lower motor neuron syndromes and motor neuropathies.
Differential diagnosis of demyelinating neuropathies, including MMN
MMN with an emphasis on the electrodiagnostic features
Pestronk, A, Choksi R, Blume, G, Lopate, G. Multifocal motor neuropathy: Serum
IgM binding to a GM1-ganglioside containing lipid mixture but not to GM1 alone.
Neurology 1997;48:1104-1106.
Pestronk A, Lopate G, Kornberg AJ, et al. Distal lower motor neuron syndrome with
high-titer serum IgM anti-GM1 antibodies -improvement following immunotherapy with
monthly plasma exchange and intravenous cyclophosphamide. Neurology
1994;44:2027-2031.
Treatment for lower motor neuron syndromes and MMN using lower cumulative doses
of intravenous cyclophosphamide.
Detailed description of different lower motor neuron syndromes and associated
autoantibodies.
Pathogenic effects of anti-GM1 antibodies.
Return to
MMN
Return to
Motor Syndromes
Return to
Neuromuscular
Home Page
9/27/98