Home, Search, Index, Links, Pathology, Molecules, Syndromes,
Muscle, NMJ, Nerve, Spinal, Ataxia, Antibody & Biopsy, Patient Info
|
Home, Search, Index, Links, Pathology, Molecules, Syndromes, Muscle, NMJ, Nerve, Spinal, Ataxia, Antibody & Biopsy, Patient Info |
 Channels & disorders Anions ATPase Calcium Cation Chloride Concepts Cyclic nucleotide-gated Gap junctions Long QT Syndromes Magnesium Mitochondrial solute carriers Na+, K+, Cl- Co-transporters Piezo Potassium HCN KCN K+/H+ ATPase Proton-gated Sodium Na+/H+ exchangers Non-voltage-gated Voltage-gated Toxins Transient receptor potential Channel binding proteins Transmitters/Receptors Acetylcholine ATP Capsaicin Catecholamines Dopamine Glutamine Glycine Purines Diagrams |
CHANNEL TYPES: General 9
|
|
Classes Voltage gated (CLCN; CLCK) Intracellular (CLIC) Calcium activated (CLCA) Anion channels (SLC4) Na-K-Cl Cotransporters Principles Disorders |
|
|
|
Figure Principles: Na+ channels Exchangers Non-voltage-gated Voltage-gated Subunits SCNA; SCNB; SCNN NAH exchangers: SLC9; SLC other Cation (CNG) Na+ channel disorders |
|
Ca++ channel disorders Ca++ channel: Figures Types Voltage-gated Ca++ channels Classes L; N; P; Q; R; T Principles CACN: A; B; G Other Ligand gated (ATP2) Intracellular activation (RYR; IP3) Ca++ sensors Cation (CNG-gated) Other (NAADP; EDG1) |
|
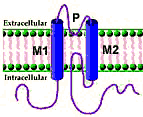
Figure K+ channel disorders Principles Structure Functions Voltage gated Inwardly rectifying KCa Subunits |
Types HCN KCN A; B; C; D; E; F; G; H; J; K; M; N; Q; S; T; V KCTD H+/K+-ATPase Plasmolipin SUR |
|
|
KCN types A; B; C; D; E; F; G; H; J; K; M; N; Q; S; T; V |
|
|
|
Group 1 TRPC (canonical) TRPV (vanilloid) TRPM (melastatin) TRPA (ankyrin) TRPN (NOMPC-like) Group 2 TRPP (polycystin) TRPML (mucolipin) |
|
| Gap junction |
| Ion channel | Binding protein | Mechanism & Effect |
|---|---|---|
|
K+ channel, Voltage-gated Shaker type NMDA receptor NR2 subunit |
Chapsyns*: PSD-95
SAP97 Sap102 |
Binding via PDZ** domains 1st & 2nd on PSD-95 Post-synaptic densities in CNS |
|
NMDA receptor
NR1 subunit |
α-actinin
|
Actin binding protein Concentrated in dendritic spines |
| Glycine receptor (GlyR) |
Gephyrin
|
Binds to β intracellular loop of GlyR & tubulin |
| AChR: Nicotinic | Rapsyn/43K | Neuromuscular junction localization |
|
Na+ channel Voltage-gated |
Ankyrin G
|
Node of Ranvier localization |
|
AMPA receptor GluR2 subunit |
Glutamate receptor interacting protein 1 (GRIP1) |
Binding via PDZ domain Couples receptor to cytoskeletal & signaling molecules |
|
Glutamate receptor Metabotropic Subunits: mGluR1a & mGluR5 |
Homer1
|
Binding via PDZ-like domain Expression ↑ by synaptic activity Cerebellar development |
|
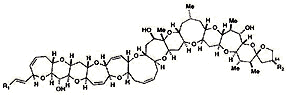
Marine toxins Ciguatoxin Conotoxins Palytoxin (Clupeotoxism) Tetrodotoxin Shell fish Saxitoxin: Paralytic Domoic acid: Encephalopathic Brevetoxins: Neurotoxic Diarrheic Other Lidocaine Potassium channel |
|
Epidemiology Toxicity Clinical Laboratory |
|
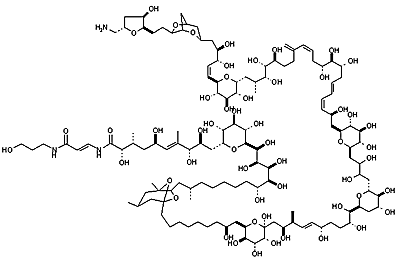 From NCI Palytoxin
|
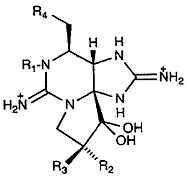
|
| Tetrodotoxin |
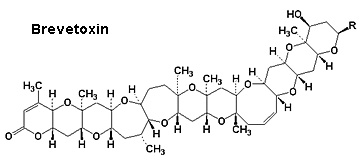 From FDA |