- Clinical syndromes: General
- Duchenne muscular dystrophy
- Epidemiology
- Incidence: 1:3,600–9,300 live male births
- Newborn screening
36:
CK levels at 12 days to 1 month = 1,800 to 5,400
- Genotype: Dystrophin
- 96% with frameshift mutation
- 30% with new mutation
- 10% to 20% of new mutations are gonadal mosaic
- Clinical
- Video
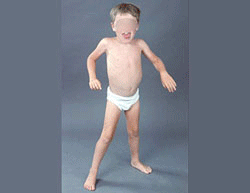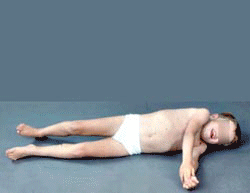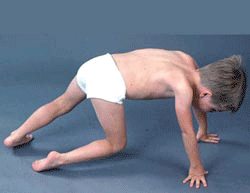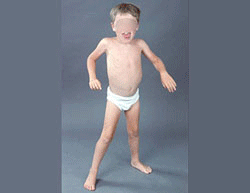
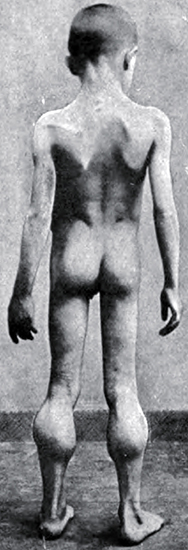
- Weakness
- Onset age: 2 to 5 yrs
- Distribution
- Proximal > Distal
- Symmetric
- Legs & Arms
- Most involved muscles: Adductor magnus in legs
- Relatively spared muscles: Gracilis & Sartorius
- Course
- Reduced motor function by 2 to 3 years
- Steady decline in strength: After 6 to 11 years
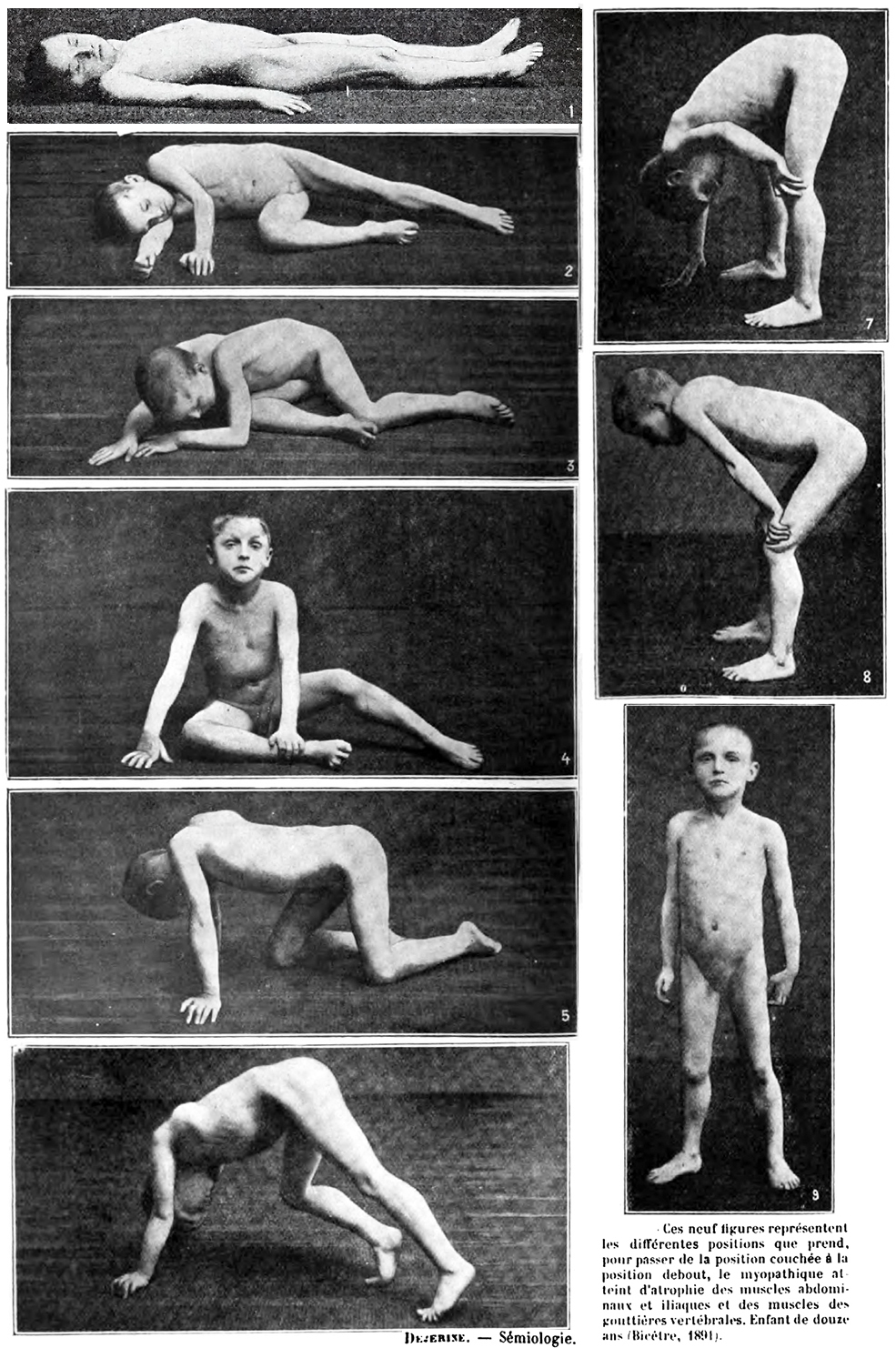
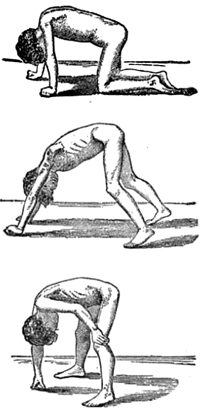
- Gowers sign
- Standing up with the aid of hands pushing on knees
- External link: YouTube
- Loss of Ambulation
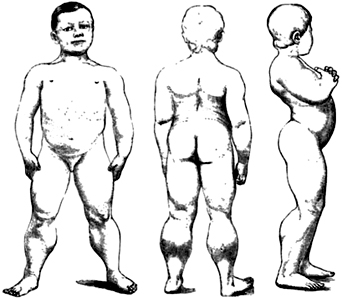
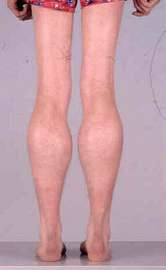
- Muscle hypertrophy
- Especially calf
- May be generalized
- Increases with age
- Most commonly due to: Muscle replacement by fat & connective tissue
- Some relatively spared muscles may have true hypertrophy
- Musculoskeletal
- Contractures
- Especially ankles; Also hips & knees
- Treatment
- Non-surgical
- Night splints on ankles: More effective than passive stretch
3
- Surgical
- Contracture release of ankles, knees or hips
- Early ambulation after surgery
- Scoliosis
- Onset: Most after loss of ambulation
- Partial relation to ambulation & growth status
- May be reduced if walking & standing are prolonged to ≥ 16 to 18 years
- Treatment: Surgical insertion of spinal rod
- Evaluation: X-ray; Sitting scoliosis series
- Timing of surgery: Usually before primary curve is 25%
- Vital capacity: Ahould be ≥ 40% of predicted
- Other clinical features
- Cardiomyopathy: Dilated; Especially > 15 years
- Mental retardation: Mean IQ ~ 88
- Night blindness
- Altered response to flashes of light in dark adapted state
- ERG: b-wave, Reduced amplitude
- Dp260: Isoform of dystrophin in retina
- Gastrointestinal: Rare
- Type: Pseudo-obstruction; Gastric dilatation
- Upper GI tract
- Late in disease course
- Death
- Most common between 15 - 25 years
- Due to respiratory or cardiac failure
- Life prolonged by ~ 6 years to 25 years with respiratory support
8
- Life shortened by 2 years with cardiomyopathy
- Laboratory
- Serum
- CK: Very high
- Highest: Ambulant patients; Up to 100x upper limit of normal
- Lower
- Increasing age: Less correlation between age & CK in Becker MD
- Lowest: Late stage disease; Severe muscle atrophy
- Birth: Increases over weeks after birth
- Troponin I: Elevated above normal but not to levels in cardiac ischemia
- Liver enzymes
18
- High AST, ALT & LDH
- Up to 23x upper limit of normal
- Levels linearly correlate with CK level
- Normal: Alkaline phosphatase, γ-glutamyl transferase
- Muscle biopsy
- Endomysial fibrosis
- Variable fiber size: Small fibers rounded
- Muscle fiber necrosis & regeneration
- Myopathic grouping
- Hypercontracted (opaque) muscle fbers
- Muscle fiber internal archetecture: Normal or immature
- Dystrophin: Absent staining
- Other membrane proteins
- Sarcoglycans: Reduced
- Aquaporin 4: Reduced; Varied levels
- Diagnostic & Lab testing
- Muscle dystrophin
- Histochemical staining: Dystrophin protein absent in muscle
- Weatern blot: Dystrophin protein absent
- Genetic: Deletion, Duplication, Small mutation, Point mutation
10
- Southern blot: Detects large deletions
- Sequencing of gene: Required to detect point & other small mutations
- MRI
- External link
- Treatments
- Prednisone
28
- Doses
- Weekly dosing
6
- Total dose: 5 to 10 mg/kg/week starting dose
- Dosing schedule: 2.5 to 5 mg/kg/day on Friday & Saturday evenings
- Side effects: Fewer than daily prednisone
- Weight gain: Some patients; Less than daily prednisone
- Cushingoid features: Mild
- Irritability: On day of dose
- Growth
- Not prominently impaired
- More growth (1" per year) than with daily prednisone
- Daily dosing
- Dose: 0.75 mg/kg/day starting dose
- Less optimal benefit:risk ratio than weekly regimen
- Effects of treatment
- Walking: Prolonged by 2 to 5 years
- Strength: Increased
- Falling: Reduced
- Pulmonary function
46
- Improved
- Benefit may continue with treatment into adulthood
- Most beneficial
- While patient still ambulatory
- ? When started at early age (3 to 5 years)
- Scoliosis: May prolong walking long enough to reduce likelihood or severity
- Deflazacort
- Dose: 0.9 to 1.2 mg/kg/day starting dose
- Benefit: Improved strength
- Side effects
- Weight gain: Frequent; Less than daily prednisone
- Vertebral fractures: High frequency; Up to 100% by 9 years
- Short stature
27
- Vamorolone
45
- Dose: 6 mg/kg/day
- Benefit: Improved strength
- Side effects
- Less growth inhibition than prednisone
- BMI increase
- Steroid insufficiency: Treat with prednisone
- Antisense oligonucleotides
- Exon 51 skip (Eteplirsen)
- Genetic effect: Change out-of-frame to in-frame deletion
- Dystrophin effect: Mild increase
- Frameshift deletions with possible benefit
- General: Must include exons 50-51 or 51-52
- Specific deletions: Exon 50; 52; 43-50; 45-50; 47-50; 48-50; 49-50
- External link: Skipping tool
- Clinical effects
- Slower decline in respiratory & arm function
- May not benefit cardiac function
- Exon 51: ExonDys-51
- Exon 53: VyonDys-53; Viltolarsen
- Exon 45: AmonDys-45
- Gene therapy
40
- Delandistrogene moxeparvovec (Elevidys)
- Protein delivered: Truncated 138 kD dystrophin
- Benefit: Probable strength stabilization
- Side effect: Immune reaction to delivered dystrophin protein
- Increased risk: Deletion mutations in Exons 1-17 or 59-71
41
- Trials
33
- Gene replacement
- Minidystropphin
- Microdystrophin
34
- Dystrophin expression: Present in muscle fibers
- Clinical: Functional improvement
- Serum CK: Reduced
- Transgene exons
- Sarepta/Roche: 1-17; 59-71
- Genethon: 1-17; 59-70
- Pfizer: 1-13; 50-51; 56-70
- Solid: 1-11; 42-45; 57-71
- Possible immunogenic Hinge 1 region: Exons 8 to 11
- Utrophin: Up regulation
- Nonsense mutation read through: Gentamycin; Ataluren
- Myostatin or ActRIIB inhibition
- ? Oxandrolone: 0.1 mg/kg/day
- Cardiac
- Exon skippable mutations & Frequencies
- Exons: 8 (4%), 44 (8%), 45 (13%), 50 (5%), 51 (15%), 52 (3%), 53 (9%), 55 (2%)
- Exon 8 & 44 skippable mutations: Longer ambulation, especially 3 to 7 & 45 deletions
- Exon 44 skippable mutations: More revertant fibers in muscle
- Exon 51 skippable mutations: Shorter ambulation
- External link: Duchenne biography
|
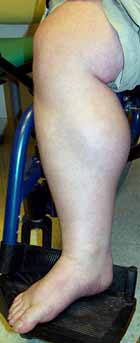
DMD: Hypertrophic
leg muscle
|
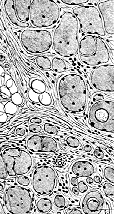
Dystrophic
muscle (Erb)
|
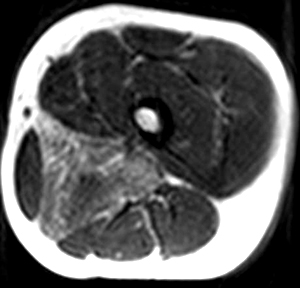
DMD MRI: Adductor magnus involvement
|
|