|
Home Index Search Links Pathology Molecules Syndromes |
Muscle NMJ Nerve Spinal Ataxia Antibody & Biopsy Patient Information |
CONGENITAL MYOPATHIES & WEAKNESS 200
Congenital Weakness: General
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

Central Core Disorders ±
Malignant Hyperthermia
 170
170
●
Ryanodine Receptor 1 (RYR1; RyR)
|
RYR1 Genetics RyR protein Clinical Laboratory Pathology Variants RyR clinical syndromes Dominant (CMYO1A) Central core diseases Congenital myopathy Cores, Rods & Malignant Hyperthermia Limb-Girdle Syndrome: Onset in teens Minicores, Transient Fetal akinesia Malignant Hyperthermia: Onset in teens King-Denborough Syndrome No cores Type 1 fiber predominance Congenital (CNMDU1) Hypotonia, Benign Exercise intolerance; Tubular Aggregates Axial myopathy, Adult onset Serum CK high: Asymptomatic Recessive (CMYO1B) Dusty Core Central core diseases Minicores, Transient Fetal akinesia Mild phenotype & Recessive inheritance Samaritan myopathy Nemaline rods + External ophthalmoplegia Congenital myopathy Centronuclear myopathy Fatigable weakness Focal lost striations Hypotonia & Cardiac Periodic Paralysis |
Cores: Variants Multicore/Minicore RYR1 SEPN1 ACTN2 CCDC78 FXR1 MYH2 MEGF10 TTN Core-Rod RYR1 NEB ACTA1 KBTBD13 (NEM6) CFL2 (NEM7) TRIP4 TNNT1 Cores: Other MYH7 Myopathy + Myofibril Δ: PYROXD1 Eccentric core: UNC45B |
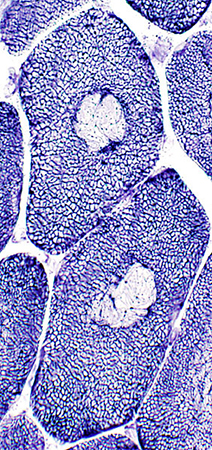 NADH Stain |
- History
- Described by: Shy & Magee
- Nosology: Central core disease, Dominant; CMYO1A; CMYP1A
- Epidemiology
- Pediatric point prevalence: 1:90,000
- Ryr1: Genetic features
11
- 106 exons
- > 700 mutations identified
- Central core families: Most linked to Ryanodine receptor mutations
- Mutation types
- Most: Missense
- Few: Small deletions
- Rare: Splice or Insertion
- Dominant mutations (CMYO1A)
- Most: Missense
- Location: Hotspots on gene
- de novo: May cause
- Core-rod myopathy
- Ser4028Leu Recurrent mutation: Hypotonia & Benign course, No cores
- Dominant variant disease mechanisms: Sarcoplasmic reticulum Ca++ release dysfunction
- RyR1 channel hyper/hyposensitivity
- Ca++ leak: Chronic
- Dominant Mutation hot spots
- Cytosolic region of protein
- Exons 2 - 19
- Cytoplasmic N terminus: Malignant hyperthermia phenotype mutations in this region
- Central cytoplasmic region: Gene segment 6400-6700
- Mutations often occur at CpG dinucleotide sequences on coding or anti-sense DNA strand
- Exons 39 - 47
- 3' Transmembrane domain (Exons 85-105): 30% of cases
- Malignant hyperthermia by IVCT
- But no clinical complications with exposure to MH triggering agents
- Cytosolic region of protein
- Dominant Mutations: Other
- 2 different mutations in codons 614 (Same as pig 615), 2163, 2454 & 2458
- 1 Mutation in C-terminus: Severe phenotype
- Dutch allele with 3 different mutations: Ile1571Val, Arg3366His, Tyr3933Cys
159
- Heterozygous: MH susceptbility
- Compound heterozygous with Val4849Ile: MH + Myopathy with cores & rods
- Irish/English/French: G341R
- French MH susceptibility: R614C
- Libyan Jews: c.12815_12825del; Dominant or Recessive syndromes
- Spanish family: Leu2286Val; Dominant; Myalgia, Cramps, CK high
- Quarter horses, Male: R2454G; Fatal anesthesia-induced malignant hyperthermia (MH) & rhabdomyolysis with hyperthermia
- Recessive mutations (CMYO1B)
- Present along entire gene
- More common with severe phenotype
- Large deletions reported: Compound heterozygous
- 54 of 106 RYR1 exons: Congenital lethal myopathy with atypical histopathologic features
- Exon 91 to 98: Recessive late-onset core myopathy
- Exons 70-71: Arthrogryposis multiplex congenita, Severe
- Recessive variant disease mechanisms
- Hypomorphic RyR1 protein expression
- RyR1-Dihydropyridine (DHPR) misalignment
- Tissue-specific epigenetic silencing of maternal RYR1 allele: Recessive core myopathies
- Clinical-Genetic correlations
- Central core disease (CCD), Dominant mutations
- C terminal region of RYR1, Missense mutations: Typical CCD; Early onset, Cores well-formed
- Outside C terminal region: Clinically Milder; Atypical cores
- Central core disease (CCD), Recesive mutations
- Mutation locations: All through coding sequence
- Phenotypes: Varied
- Severe
- Ophthalmoplegia + Face weakness
- Pathology: Multiminicores; Central nuclei; Fiber-type disproportion
- Muscle fibers: Internal nuclei
- MH alone mutations: Most in N-terminus region
- Arg614Cys: Common (4% to 9%)
- Also produces porcine MH
- Higher thresholds & Smaller contractures compared to other mutations
- Gly2433Arg: Common (4% to 7%)
- Gly341Arg: 5% to 6% of British & Irish families
- Arg2454Cys: Low trigger thresholds
- Other: Gly248Arg; Arg552Trp; Arg614Leu; Val2168Met; Thr2206Met
- Arg614Cys: Common (4% to 9%)
- Central core disease & MH mutations
- Arg163Cys; Ile403Met; Tyr522Ser; Arg2434His; Arg2454His
- Severe & penetrant phenotype: Ile4898Thr
- Mexican pedigree
- C-terminal transmembrane/luminal region
- Channel properties
- Leaky
- Activated by Ca++ concentrations 4-fold lower than normal
- Central core disease (CCD), Dominant mutations
- Allelic disorders
- Ryanodine receptor protein
81
- Size
- 560 kDa
- Largest ion channel protein
- Location
- Sarcoplasmic reticulum, terminal cisternae
- Structure
- RYR1 activation
- Ryr1 activation by: Cav1.1 voltage sensor proteins
- Location: Transverse tubule membrane
- During Excitation-Contraction
- S-Palmitoylation
204
- 18 palmitoylated cysteine residues in RyR1 cytoplasmic domain
- Modifications stabilize RyR1 open conformation
- Enhance Ca++ release from sarcoplasmic reticulum
- Ryr1 activation by: Cav1.1 voltage sensor proteins
- RyR1 Functions
- Structural: In skeletal muscle
- Foot: Bridges gap between sarcoplasmic reticulum & T-tubule
- Calcium release channel
Excitation-Contraction Coupling: Muscle 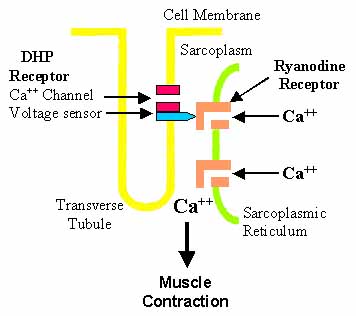
- Major Ca++ channel expressed in skeletal muscle
- Calcium release, Calcium-induced
- With Ca++ channels in plasma membrane
- Excitation-Contraction coupling
- Rise in Ca++ required for muscle fiber contraction
- Skeletal muscle Ca++ release channel
- Provided by activation of sarcoplasmic reticulum Ca++ release channels in RyRs
- Ca++ release: From sarcoplasmic reticulum stores (lumen) into cytoplasm
- Functionally coupled to changes in sarcolemmal membrane potential
- Through L-type VGCC (DHPR)
- RyR1 opening
- Induced by: Conformational changes in DHPR (Physical interaction)
- Stimulus: Membrane depolarization
- RyR2 activation in heart
- Activation of L-type Ca++ channels
- Induces Ca++-induced Ca++ release (CICR)
- Regulatory molecules
- Activation during excitation-contraction: Cav1.1 voltage sensor proteins
- Ca++
- Mg++: Inhibitory
- Adenine nucleotides & Cyclic ADPribose: Channel activation
- Oxidation
- Nitrosylation
- Caffeine & 4-chloro-m-cresol (CmC)
- Activate RyR channels
- Facilitate channel opening: ↑ Sensitivity of RyR to Ca++-dependent activation
- RyR blockers; Ruthenium red; Local anesthetics (Procaine; Tetracaine)
- Dantrolene: Inhibits SR Ca++-leak via mutant RyR1; Used to prevent MH crises
- Structural: In skeletal muscle
- Associated proteins
- Dihydropteridine receptor (DHPR)
- FK506 binding protein 1A (FKBP1A)
- Calmodulin (CaM):
Participates in Ca++-dependent regulation of channel activity
- Ca++-free CaM: Activates RyR
- Ca++-bound CaM: Inhibits RyR channel function
- Triadin
- Calsequestrin
- Phosphatases: PP1
, PP2A
- Phosphodiesterase PDE4D
- Muscle A-kinase anchoring protein (mAKAP; AKAP6)
- Stac3
- Kinases: PKA; CamK
- Mutation effects on protein function
- Common central core disease effect: Uncouple excitation from contraction
- Generate leaky RyR1 channel
- Interfere with DHPR binding
- More MHS + CCD with
- Greater Sensitivity of mutant RYR protein to agonists caffeine & halothane
- Higher resting Ca++ in sarcoplasm
- Lower endoplasmic reticulum Ca++ stores
- Reduced Size of releasable Ca++ stores
- Y522S: Leaky RyR1
- C-terminus (14895T & other): Uncouple EC coupling
- Common central core disease effect: Uncouple excitation from contraction
- Size
- Central Core Congenital Myopathy (CMYP1A
): Clinical features
- Genetics
- Inheritance: Dominant
- Mutations
- Hotspot: C-terminal exons 101�102
- C-terminal: Earlier onset; Cores more distinct
- Outside C-terminal: Milder phenotypes; Cores Atypical
- Clinical
- Onset age
- Childhood or Congenital
- May vary widely within families
- Developmental
- Fetal movements: Reduced
- Breech presentation
- Motor
- Hypotonia
- Weakness
- Proximal: Hips
- Legs > Arms
- Vastus lateralis, Adductor magnus
- Axial
- Face: Mild weakness
- Extraocular movements: Normal
- Discomfort
- Cramps
- Myalgias
- Skeletal
- Congenital hip dislocation
- Scoliosis
- Foot deformities
- Malignant hyperthermia
- Cardiac: Normal
- Course: Non- or Slowly Progressive
- Onset age
- Laboratory
- Genetics
- Variants: Central core syndromes & Other RyR disorders
- RYR1 variant: Malignant Hyperthermia, Onset in teens
- RYR1 variant: King-Denborough Syndrome
- RYR1 variant: Limb-Girdle Syndrome
- Inheritance: Dominant
- Clinical
- Onset age: Teens
- Weakness: Proximal; Symmetric
- Malignant Hyperthermia
- Course: Slowly progressive
- RYR1 variant: High serum CK: Asymptomatic
- RYR1 variant: Central core disease, Rods & Malignant Hyperthermia with RYR1 mutation
- Mutations
- Y4796C: C-terminal channel forming domain
- Thr4637Ala: Transmembrane region
- Epidemiology: 2 families
- Clinical
- Onset: Infantile
- Weakness: Non-progressive; Hypotonia; Walk at 2.5 years
- Contractures: Ankles
- Course: Still walking in 3rd & 4th decades
- Laboratory
- Serum CK: Normal
- Muscle biopsy: Central cores; Rods in areas without cores
- In vitro contracture test: Positive
- Other Rod-Core syndrome: Nebulin mutations
- Mutations
- RYR1 variant: Nemaline (Rod) Myopathy With Ophthalomoplegia, Severe Congenital
100
- Genetics
- Inheritance: Recessive
- Mutations: Pro1573Leu & Asp2529Asn
- Clinical
- Fetal akinesia
- Hypotonia & Weakness
- Neonatal
- Severe
- Generalized
- Face
- Respiratory insufficiency
- Swallowing disturbance
- Ophthalomoplegia
- Face: Narrow
- Laboratory
- Muscle histology
- Nemaline bodies: Cytoplasmic
- Type 1 fibers: Small; Predominant (70%)
- No central cores or minicores
- EMG: Myopathic; Small, short action potentials
- Muscle histology
- Genetics
- RYR1 variant: Dusty Core Syndromes
148
- Epidemiology: Common RYR1 Recessive syndrome
- Genetics
- Inheritance: Recessive
- Mutations
- Nonsense & Missense
- Especially in Bridge solenoid & Pore domains
- Clinical
- General: More severe & earlier onset vs other RYR1 recessive syndromes
- Onset ages: Median 1 year; Range = Antenatal to 66 years
- Motor milestones: Delayed; May not walk
- Eye (50%): Ptosis; Ophthalmoplegia; More with severe disease
- Weakness
- General: Often more proximal
- Face: Weak; May be long & narrow
- Arms & Legs
- Respiratory (40%)
- Skeletal: Scoliosis; contractures
- Laboratory
- Muscle
- Muscle fiber lesions
- Irregular areas of myofibrillar disorganisation
- Gomori trichrome: Red-purple granular deposition
- Uneven oxidative stains: Clear center; Radial strands
- ATPase stain: Reduced or Absent in center of fibers
- Central cores (10%)
- Desmin, Myotilin, RYR1, DHPR & αB-crystallin: Central staining
- May be more prominent with increasing age
- Type 1 fiber predominance
- Internal nuclei
- Fiber sizes: Varied
- RyR1 expression: Lowest levels
- Ultrastructure
- Sarcomere disorganization
- Osmophilic filamentous smeared material
- Short thickened darker fragments
- Muscle fiber lesions
- Muscle
- RYR1 variant: Central core disease with transient Minicores (CMYO1B)
30
- Nosology
- Multicore syndrome: Mild phenotype
- Multicore syndrome: "Central core-like"
- CMYP1B
- CMYO1B
- Epidemiology: Algerian, Turkish & German families
- Genetics
- Inheritance: Recessive
- RYR1 mutation: Homozygous; Missense; P3527S
- Overlap with phenotype for central core syndrome: Missense V4849I
- Clinical
- Onset
- Age: Infancy or childhood
- Hypotonia
- Weakness
- Severity
- Usually Moderate
- Variable in families: Some patients with severe disability
- Distribution
- Proximal > Distal
- Axial muscles: Neck; Trunk flexors
- Pelvic girdle
- Hands: Moderate weakness; Wasting
- Face: Some patients
- Respiratory
- Vital capacity variably reduced: 50% to 100% of normal
- No respiratory failure
- Course: Stable or Slowly progressive
- Severity
- Other muscle
- Preserved bulk
- Pain on exertion
- Ophthalmoplegia: Some patients
- Joints
- Hyperlaxity: Hand involvement; Other joints also involved; Patellar, Hip & Knee dislocations
- Contractures: Some patients; Mild; Mainly at heels
- Arthrogryposis: Some patients
- Onset
- Laboratory
- Serum CK: Normal
- Cardiac: Normal
- MRI: Gluteus maximus & Quadriceps preferentially involved
- Muscle biopsy
- Childhood: Multiple minicores
- Minicores or Cores: Larger than with SEPN1 mutations
- Adult: Central cores; Muscle fiber loss
- Type 1 muscle fiber: All or Predominance
- Myopathic changes
- Nosology
- RYR1 variant: Central core disease with Fetal Akinesia (FADS)
45
- Epidemiology
- Frequency: Up to 8% of FADS
- Genetics
- Inheritance: Dominant or Recessive
- Mutations
- Recessive
- R614C (Often associated with MH) & G215E
- L4650P & K4724Q
- In-frame deletion: c.2097_2123del
- Nonsense: c.6721C>T
- Dominant: G4899E (Mother less severely affected)
- Recessive
- Clinical
- Pregnancy
- Fetal akinesia
- Differential Dx in NM disorders: Nemaline & Myotubular myopathies; Pena-Shokeir
- Fetal movements: Reduced
- Arthrogryposis: Multiple
- Hydramnion
- Fetal akinesia
- Post-natal
- Hypotonia: Severe
- Respiratory distress
- Multiple malformations
- Course
- Death in some
- Others with stable weakness after prolonged respiratory support
- Ocular: Strabismus & Ptosis may develop
- Pregnancy
- Muscle pathology
- Central cores: Eccentric; Some patients
- Fiber size: Varied
- Type 1 fibers: Small
- Central nuclei: Some patients
- Endomysial connective tissue: Increased
- Fiber types: May be uniform & abnormal
- Ultrastructure: Myofibrillar disorganization
- Epidemiology
- RYR1 variant: Central core disease: Mild phenotype & Recessive inheritance
- RYR1 Genetics
- Missense V4849I mutation
- Same mutation also found in dominant malignant hyperthermia family
- Disease pattern overlaps with mild phenotype of multicore disease
- RYR1 Genetics
- RYR1 variant: Congenital neuromuscular disease with uniform type 1 fibers (CNMDU1)
69
- RYR1 genetics: Mutations
- Inheritance: Dominant or Recessive
- Heterozygous
- Missense or Deletion
- Location
- Common: C-terminal domain of RYR1; Pore forming segment
- Other: Arg2615Cys; Arg4893Trp
- Mutations found in 40% of patients with CNMDU1
- None in patients with mental retardation
- Clinical
- Onset: Congenital or Childhood
- Weakness
- Mild to Severe
- Proximal ± Axial
- Face: Some patients
- Progressive
- Tendon reflexes: Reduced or Absent
- CNS: No mental retardation
- Laboratory
- Serum CK: Normal
- EMG: Myopathic
- Muscle biopsy
- Fiber type: All, or most, type 1
- No cores
- Endomysial connective tissue: Normal or Mild increase
- RYR1 genetics: Mutations
- RYR1 variant: Benign Samaritan congenital myopathy
92
- Epidemiology: Samaritan Israeli family, inbred
- Genetics
- Inheritance: Recessive
- RYR1 Mutation: Homozygous; Missense; Tyr1088Cys
- Clinical
- Onset
- Age: Congenital
- Hypotonia: Severe
- Weakness
- Birth: Severe; Diffuse; Respiratory failure; Hypotonia
- Improves with age
- Adults: Mild proximal weakness in arms & legs
- Eye movements: Normal
- Face: Dysmorphic
- Hypertelorism
- Epicanthal folds
- Nasal bridge: Broad
- Mouth: Small
- Hyperthermia with anesthesia: Mild in 1 patient
- Onset
- Laboratory
- Serum CK: Normal
- EMG: Mild myopathy
- Muscle pathology
- Internal nuclei
- Core-like structures: In type I muscle fibers
- Fiber types: Types I & II present
- Internal architecture: Irregular; Moth-eaten
- RYR1 staining: Reduced; Irregular; Aggregates with Caveolin-3
- Central core disease or Multicores
- Genetics: Missense mutation Arg 4893Trp; Heterozygous
- Dominant inheritance
- RYR1 variant: Congenital myopathy with focal loss of cross-striations
98
- Genetics
- Inheritance: Recessive
- RYR1 mutations: Heterozygous; Arg3539His, c.10627-2A>G splice site
- Clinical
- External ophthalmoplegia: No ptosis
- Motor development: Delayed
- Weakness: Generalized; Proximal > Distal
- Progression: Minimal
- Contractures: Long finger flexors
- Muscle
- NADH stain irregular
- Type 1 predominance
- Genetics
- RYR1 variant: Bent spine syndrome (Axial myopathy, Adult onset)
101
- Genetics
- Inheritance: Dominant
- RYR1 Mutations: Heterozygous; Missense; Varying gene regions
- Clinical
- Onset ages: 3rd to 7th decade
- Weakness
- Axial
- Lumbar: Lumbar hyperlordosis
- Neck
- Shoulder fixators (Scapular winging)
- Camptocormia 10%
- Proximal limb: Mild
- Axial
- Myalgia: Common; Exercise related
- Laboratory
- Serum CK: High; Up to 1,400
- Malignant hyperthermia testing: Positive in 1 patient
- Muscle
- Varied fiber size
- Internal nuclei
- Internal architecture: Irregular
- Cores: 10% of patients
- Type 1 predominance: 10%
- Muscle imaging: Lumbar paraspinal & Posterior thigh abnormal
- Genetics
- RYR1 variant: Rhabdomyolysis/Exertional myalgia
107
- Genetics: RYR1 mutations
- Heterozygous
- Missense: Some previously associated with malignant hyperthermia
- Clinical
- Onset age: 3 to 45 years
- Rhabdomyolysis (80%)
- Exertional myalgia, isolated (20%)
- Ptosis
- Strength: Normal
- Occasional features: Heat intolerance; Cold-induced muscle stiffness
- Laboratory
- Resting CK: Normal to 1100
- Muscle biopsy
- Fiber size: Varied
- Internal nuclei
- Internal architecture: Irregular or Cores
- Genetics: RYR1 mutations
- RYR1 variant: Congenital myopathy + Fatigable weakness
112
- Epidemiology: 2 families, 3 patients
- Genetics
- Inheritance: Recessive
- Mutations: Arg2241X nonsense; Arg109Trp, Leu2963Pro missense
- Clinical
- Onset
- Age: Congenital
- Hypotonia
- Ptosis, Fatiguable
- Myopathic facies
- Feeding difficulties
- Weakness
- Proximal
- Respiratory
- Walking: Delayed
- Course: Progressive
- Axial hypotonia
- Eyes: Ophthalmoparesis, Ptosis
- Skeletal
- Scoliosis
- Achilles tendon contractures, bilateral
- Malignant hyperthermia: Susceptibility
- Treatment: Pyridostigmine
- Onset
- Laboratory
- Serum CK: Normal
- Muscle ultrasound: Echogenicity increased
- Muscle pathology
- Internal nuclei
- Fiber size: Atrophy
- Type I fibers: Smaller; Predominant
- Core-like areas: Rare
- EMG: Myopathic
- Muscle (thigh) MRI
- Symmetrical involvement
- Muscles: Biceps, Semi-membranosus, Vasti & Adductors
- RYR1 variant: Hypotonia + Benign course
172
- Epidemiology: 13 patients, 9 families
- Genetics
- Inheritance: Dominant or de novo
- Mutation: c.12083C>T (p.Ser4028Leu)
- Clinical
- Onset age: Pre-natal to Childhood
- Hypotonia
- Weakness: Proximal & Trunk ± Diatal or Facial
- Symptoms: Fatigability; Myalgia
- Skeletal: Joint hyperlaxity; Scoliosis
- Course: Early improvement
- Laboratory
- Muscle pathology: Type I predominance; Fiber sizes varied
- Serum CK: Low
- RYR1 variant: Centronuclear myopathy
142
- Epidemiology
- > 35 patients
- Common geographic region: South Africa, South America
- Genetics
- Inheritance: Recessive or Sporadic
- Mutations: Compound heterozygote; Missense > Splice, Frameshift
- Clinical
- Onset age: Congenital or Pre-Natal
- Early
- Fetal movements: Reduced
- Hypotonia
- Feeding difficulties
- External ophthalmoplegia
- Weakness
- Proximal ± Axial > Distal
- Especially: Biceps; Quadriceps; Hip abductors
- Bulbar: Dysphagia (75%)
- Neck flexion (75%)
- Face: Myopathic facies; Inverted V-shaped mouth
- Maximal motor function: Sitting or Walking
- Respiratory: Ventilator dependent; Frequent respiratory infections
- Proximal ± Axial > Distal
- Skeletal
- Spine: Scoliosis (30%); Rigid spine (30%)
- Contractures (35%)
- Cardiac: Normal
- Course: Slow improvement or Non-progressive
- Muscle MRI
- Involvement of: Vastus lateralis, Adductor magnus, Biceps brachii
- Muscle pathology
- Nuclei: Central (20% to 90% of fibers); Internal, often multiple
- Type 1 muscle fibers
- Predominance
- Size: < Type 2
- Cores & Mini-cores: May develop with increasing age
- NADH
- Central increase
- Peripheral halos: Few patients
- No necklace fibers or radial strands
- Ultrastructure: Z-line streaming; Core-like irregularities
- Epidemiology
- RYR1 variant: Periodic Paralysis (Episodic weakness) ± Myopathy
- Epidemiology: 4 patients
- Genetics
- Mutation types: Misense & Stop; None with 2 stop mutations
- Mutation number: 2 or 3 in 3 patients; Single (Heterozygous) in 1 patient (Arg1043His mutation)
- Mutations: Asp708Asn, Arg2241X, Arg2939Lys; Gln70X, Arg109Trp, Met485Val; Arg1507Gln, Gly2446Ser
- Inheritance: Recessive
- Clinical
- Onset ages: Neonatal to 23 years
- Weakness, Fixed: 2 patients
- Proximal & Trunk
- Weakness, Episodic
- Limbs ± Ophthalmoplegia & Face
- Duration: Hours to Days
- Associated features: Myalgias or Cramps
- Morphology: Thin face
- Laboratory
- Serum CK: Normal
- EMG: Myopathic or Normal
- Muscle biopsy
- Fiber size: Varied
- Intermal nuclei
- Core-like structures: 2 patients
- Type 1 predominance: 1 patient
- RYR1 variant: Congenital Hypotonia & Cardiac Disorders
173
- Epidemiology: 1 patient
- Genetics
- Inheritance: Recessive
- Mutations: Intronic; c.6274+1G>A, c.10441-48G>A
- Clinical
- Onset: Neonatal
- Hypotonia
- Movements: Reduced
- Tendon reflexes: Reduced
- Ventilator dependent
- Survival: < 1 week
- Laboratory
- NCV: Axon loss
- Serum CK: Normal
- Muscle MRI: Atrophy of thigh & abdominal muscles
- Muscle pathology
- Fiber sizes varied: Type 2 small
- 2C fibers: Many
- RYR1 staining: Reduced
- Ultrastructure: Sarcomere organization: Abnormal
- Brain MRI: CNS hemorrhages
- Circulatory: Transposition of great arteries
- Management
- Avoid anesthesia-related malignant hyperthermia
- Albuterol: May improve weakness
- RYR1 variant: Exercise intolerance, High CK & Tubular aggregates
196
- Epidemiology: 2 patients
- Genetics
- Inheritance: Dominant
- Mutations: Missense; Bridging solenoid domain; Thr2206Met, Gly2434Arg
- Clinical
- Onset age: Child to early adult
- Muscle stiffness: With Exercise & Cold exposure
- Strength: Normal
- Pigmenturia: With effort
- Laboratory
- Serum CK: High
- EMG: Myopathic of Normal
- Muscle pathology
- Tubular aggregates: In type 2 fibers; Often subsarcolemmal; Multiple in single fibers
- Fiber sizes: Varied
- Type 2 fiber predominance
- Laboratory
- CK
- High in central core & MH patients
- CK may be high in some asymptomatic carriers (G341R)
- In vitro contracture test for malignant hyperthermia
- Sensitivity: 97% to 99%
- Specificity: 78% to 94%
- CK
- Muscle MRI
52
- Features of muscle involvement
- Muscle signal: Increased T1
- Clinical correlation: Similar patterns with different clinical syndromes & RYR1 mutations
- Thigh
- Involvement: Vasti, Sartorius, Adductor magnus
- Spared: Rectus, Gracilis, Adductor longus
- Lower leg
- Involvement: Soleus, Gastrocnemius, Peronei
- Spared: Tibialis anterior
- Features of muscle involvement
- Central core: Muscle pathology
- Myopathy
- Muscle fiber size: Variable
- Endomysial connective tissue: Increased
- Internal nuclei: More in older patients
- Cores: Internal zone, "core", in muscle fiber
- Reduced, absent, or abnormal in core
- Oxidative enzyme activity
- Mitochondria
- Central myofibrillar structure: Some cores
- Z-disk morphology
- Core staining: Most prominent internal pathology on
- NADH
- Mitochondrial stains
- Core length
- Millimeters to Whole muscle fiber: Longer with C-terminal RYR1 mutations
- Core location
- Central in muscle fiber
- More central with: More severe disease; Older age
- Core number
- Usual: 1 per muscle fiber cross-section
- Mutations away from C-terminus may have: Multiple small core-like structures (Mini-cores)
- Core structure types
- Structured
- Central region with: Striated myofibrillar pattern; ATPase staining
- Disease inheritance: Dominant
- Gene: RYR1
- Unstructured
- Central region with: Reduced ATPase staining
- Disease inheritance: Recessive
- Gene: RYR1
- Atypical
- Pattern
- Borders: Indistinct
- Location in fiber: Periphery/Subsarcolemmal
- Shape: Ovoid
- Number: Often > 1 per fiber
- RYR1 Mutation locations: Non-C terminal
- Pattern
- Multi-Minicores
- Dusty cores
- Gene: RYR1, Recessive
- Core-Rods
- Absent cores + Type 1 muscle fiber predominance
- May occur in younger patients
- Structured
- Core development
- Fiber type predominance may precede core formation
- Cores may start at ends of fibers & extend centrally
- Increased age: More central cores
- Proteins in cores
36
- Myofibrillar proteins
- αB-Crystallin; Desmin; Filamin C, Myotilin
- Similar patterns of immunoreactivity seen in targets
- Ca++-related proteins
- Depleted from the cores: TOM20
- Present or Reduced: RYR1
- Accumulate in, or around, lesions
- Other sarcoplasmic reticulum proteins: Calsequestrin, SERCA1/2 & Triadin
- T-tubule protein: Dihydropyridine receptor-α1-subunit (DHPR)
- SelN mutation-related minicores: Normal distribution of Ca++-related proteins
- Other: Small heat-shock proteins
- Myofibrillar proteins
- Reduced, absent, or abnormal in core
- Fiber types: Single type in most
- ATPase stain: Marked type I muscle fiber predominence
- When both fiber types present: Cores tend to be in type I fibers
- Myosin heavy chain: Often contain both type I (MYH7) & type IIX (MYH1)
- Clinical correlations
- Well formed cores + Marked type 1 predominance
- Morphology: Round; Single; Central
- Myopathy syndrome: Often without MHS
- RYR1 mutations: 90% with 60% in C-terminus
- Variable cores + Less type 1 predominance + Internal nuclei
- Morphology: Indistinct borders; Peripheral or Subsarcolemmal; Ovoid; Multiple
- MHS syndrome: Usually normal strength
- RYR1 mutations: 35%, Outside C-terminus
- Well formed cores + Marked type 1 predominance
- Myopathy
- RyR: Other
- Antibodies vs RyR1
- RyR2 mutations
- Catecholaminergic polymorphic ventricular tachycardia (CPVT)
- R176Q mutation: Arrhythmogenic right ventricular dysplasia (ARVD)
Nemaline (Rod) Myopathies
|
Rods: General Clinical features: General Laboratory features: General Pathology Rods: Hereditary NEM1: α-tropomyosin 3 (TPM3); 1q21; Dom or Rec NEM2: Nebulin (NEB); 2q23; Recessive NEM3: α-Actin (ACTA1); 1q42 Dominant Recessive NEM4: β-tropomyosin(TPM2); 9p13; Dom or Rec NEM5: Troponin T1 (TNNT1); 19q13; Dom or Rec NEM6: KBTBD13; 15q22; Dominant NEM7: Cofilin-2 (CFL2); 14q13; Recessive NEM8: KLHL40; 3p22; Recessive NEM9: KLHL41; 2q31; Recessive NEM10: LMOD3; 3p14; Recessive NEM11: MYPN; 10q21; Recessive NEM: Ryanodine receptor (Ryr1); 19q13 Rods + Cores: Dominant Rods + Ophthalmoplegia: Recessive NEM: RYR3; 15q13; Recessive NEM + Cardiac: CAP2; 6p22; Recessive Arthrogryposis (DA2B2): TNNT3; 11p15 MPD5: ADSSL1; 14q32 Also Cap myopathy CFTD Filamin C Klippel Feil 4: MYO18B MFM7 Myopathy with Myofibril Δ: PYROXD1 Pyruvate carboxylase deficiency Rod myopathy, mild Rods + Filaments: MYH2; 17p13 Zebra body |
Rods + Cores ACTA1 NEB KBTBD13 CFL2 RYR1 TRIP4 TNNT1 Rods: Sporadic disorders Infant onset myopathy Adult onset myopathy (SLONM) HIV rod myopathy Rod Myopathy Distal Weakness & Contractures  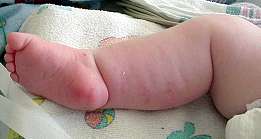 From: A Connolly |
Rod myopathies: General features 22
- Epidemiology
- Genetics
- Inheritance: Dominant, Recessive & Sporadic
- Common mutations: Skeletal muscle α-Actin (ACTA1); Nebulin
- Similar phenotype
- Variable features from single genes
- Dominant or Recessive inheritance: α-Actin; α-tropomyosin 3 (TPM3); TNNT1
- Lethal congenital or Late onset: α-Actin; α-tropomyosin 3 (TPM3)
- Mutant proteins
- Associated with structure or regulation of Thin filaments
- Mutations in same spectrum of genes also produce
- Cap myopathy
- Congenital fiber type disproportion: Size or Frequency
- Clinical features
- Onset
- Overall: Congenital (90%) to Adult
- Severity: Varied
- Early onset patients
- Respiratory failure & Feeding difficulties common: May be out of proportion to limb weakness
- Motor
- Early: Hypotonia
- Weakness patterns
- Degree: Varied
- Severe in 40% to 50%
- Most < 50% of normal strength
- Other
- Mild or Moderate severity
- Distal Predominant
- Bulbar dysfunction: Common
- Dysphagia (60%): Especially ACTA1
- Respiratory (60%)
- Most severe: Neck flexors; Hip girdle
- Distal: Nebulin, ACTA1, TNNT3, TPM2
- Ophthalmoplegia: CFL-2; KLHL40
- Paraspinous or Posterior neck: Adult onset
- Degree: Varied
- Slow movements: KBTBD13
- Ophthalmoplegia: LMOD3
- CNS: Cognitive involvement in younger onset patients
- Cardiac
- GI: Acid reflux (50%)
- Skeletal
- Contractures
- Arthrogryposis: Severe congenital cases
- Mildly reduced joint motion: 75%; May be progressive
- Scoliosis: 70%; Often progressive in late 1st decade
- Bone fractures: 30%
- Klippel-Feil: MYO18B
- Contractures
- Course
- Weakness
- Often stable
- Progressive: TNNT1, Recessive
- Morbidity from respiratory infections & feeding problems less with increasing age
- Walking
- Rare in congenital rod myopathy
- Childhood onset usually remain ambulant
- Weakness
- Mortality
- Deaths due to respiratory insufficiency
- Early increased with
- Respiratory insufficiency
- Arthrogryposis
- Failure to achieve early motor milestones
- Onset
- Laboratory
- CK: Normal (90%) or ± Elevated in adult-onset forms
- EMG: Myopathic or Neuropathic (Especially severe cases)
- Muscle pathology: Thin filament disorders
- Rods
- Histochemical appearance
- Dark red-blue structures
- Visible on
- Subcellular location: Originate from Z-disks
- Myopathy: Tend to cluster under sarcolemma & around nuclei
- May be less prominent: Infants
- Differential diagnosis: Cytoplasmic bodies
- Size: 1 to 7 μm in length
- Frequency: Often increase in number with age
- Composition
- Ultrastructure
- Electron dense
- Rod shaped
- Emanate from Z-lines
- Thickened Z-lines, or
- Extend along long axis of thin filaments
- Distinctive features
- Underlying pathogenic process
- Rod formation 2° to contractile dysfunction
- Load-dependent processes may be involved
- Rods: Differential diagnosis
- Sarcoglycanopathies
- Target fibers (Denervation): Usually Cytoplasmic bodies, not rods
- Tenotomy: Usually cytoplasmic bodies, not rods
- Neostigmine myopathy 23
- HIV-associated myopathy: Usually Cytoplasmic bodies, not rods 24
- Also see: Cytoplasmic bodies
- Histochemical appearance
- Endomysial connective tissue
ROD MYOPATHIES: Specific syndromes
NEM1 Rod myopathy
● α-Tropomyosin 3 (TPM3)
- Nosology: CMYP4B; CMYO4B; NEM1
- Epidemiology
- Infrequent cause of nemaline rod myopathy
- Frequency: < 3%
- TPM3 Genetics
- Inheritance: May be Dominant or Recessive
- Mutation hotspot: Arg168 (Arg168His, Arg168Cys, Arg168Gly)
- TPM3 Clinical-Genetic correlations
- Mildest syndromes: Missense mutation; Dominant inheritance
- Severe syndromes: Stop codon; Recessive inheritance
- Tropomyosin function
- Allelic disorders
- TPM3 protein
- Muscle: Expressed in type I muscle fibers
- 11 different forms: Expressed in many tissues including brain
- Actin-binding tropomyosin family
- Component of sarcomeric thin filament troponin-tropomyosin complex
- Predominantly expressed in type 1 (slow twitch) muscle fibers
- Regulates: Muscle contraction; Binding of myosin head to actin filament
- See: Muscle fiber proteins
- CMYO4A: NEM 1 Rod myopathy, Dominant inheritance
- TPM3 Mutations: Missense; Met9Arg, Arg167His 38
- Onset: 0 to 15 years
- Clinical
- Weakness
- Development: Motor delay in some patients
- Distribution
- Symmetric
- Distal Legs (Ankle dorsiflexors)
- Arms
- Respiratory failure
- Proximal weakness: Later in disease course
- Progression: Slow; Wheelchair often by 40 years
- More severe than TPM2 mutations
- Tendon reflexes: Reduced
- Achalasia: Involvement of smooth muscle
- Morphology
- Kyphoscoliosis
- Slender build
- Long face
- Weakness
- Laboratory
- Serum CK: Normal
- Muscle MRI: 2 patterns
- Posterior: Thigh & Legs
- Sartorius & Adductor magnus
- Pathology
- Type I fibers: Atrophy; Predominance
- Rods: In Type I fibers; May be infrequent
- CMYO4B: NEM1 Rod myopathy, Recessive inheritance
- Early onset, severe disease
- TPM3 Mutation: Homozygous; Stop at codon 31
- Onset: Birth
- Motor development: Extremely delayed and impaired
- Cognitive: Appropriate for age
- Death: 21 months
- Pathology
- Rods: Only in type 1 fibers
- Type 1 muscle fiber smallness
- Loss of α-tropomyosin 3 from muscle
- Intermediate syndrome35
- Family history: Sporadic
- Genetics
- Compound heterozygote
- Mutations: X285Ser; Splice site at skeletal muscle-specific translational stop signal
- Mutated protein: contains 57 additional amino acids
- Clinical
- Onset: Birth; Hypotonia
- Motor
- Walking: 18 months
- Wheelchair: 6 years
- Weakness: Proximal
- Face: Long & narrow; High arched palate
- Muscle pathology
- Rods: In type I muscle fibers
- Endomysial connective tissue: Increased
- Type I fiber predominance
- Internal nuclei
- Small fibers both types
- Reduced β-tropomyosin in muscle fibers
- Early onset, severe disease
- TPM3 Variant syndrome: Congenital fiber type disproportion
70
● Dominant or Sporadic - General
- Most common cause of CFTD: ~20% to 25%
- Type 1 fibers: At least 50% smaller than type 2
- Type 2 fibers: Normal or large sized
- Genetics
- Mutations: Missense; Heterozygous
- Locations: Leu100Met, Arg168Cys, Arg168Gly, Arg168His, Lys169Glu, Glu174Ala, Arg245Gly
- Arg168Cys mutation: May be associated with CFTD & nemaline myopathy pathology in same family
- Clinical
- Onset
- Age
- Usually < 1 year
- Variable: Even within families
- Hypotonia
- Poor head control
- Age
- Weakness
- Proximal > Distal
- Legs > Arms
- Neck: Flexion & Extension
- Ankle: Dorsiflexion
- Trunk: Paraspinal; Abdominal
- Facial: Mild; Myopathic face
- Ptosis: Mild; Unilateral or Bilateral
- Respiratory
- Frequency: 90%
- Nocturnal noninvasive ventilation: Onset 3 to 55 years
- Even in some ambulatory patients
- Scapular winging: Mild; 40%
- Quadriceps: Relatively preserved
- Course
- Improvement in strength & function over time
- Patients often eventually walk or run
- Other muscle
- Amyotrophy: Generalized
- Body habitus: Thin
- Gait: Waddling; Foot drop
- Skeletal
- Young children: Hypotonic stance; Exaggerated lumbar lordosis & thoracic kyphosis
- Late childhood or adulthood: Mild-to-severe kyphoscoliosis with neck extensor contractures
- Other contractures: Uncommon
- Possible symptomatic treatments
- Pyridostigmine
- 3,4-Diaminopyridine
- Onset
- Laboratory
- EMG: Normal or Myopathic
- Single fiber EMG: Abnormal jitter without blocking
- NCV: Normal
- Cardiac: Normal
- Muscle MRI
- No selective muscle involvement
- Interstitial connective tissue: Mildly increased
- Muscle pathology
- Type 1 muscle fibers
- Small size
- 60% of normal; Range 30% to 80%
- 23% to 50% of Type 2 size
- Predominance: Minority of patients
- Small size
- Type 2 muscle fibers
- Size: Large; 160% of normal; Range 110% to 220%
- Type 2B: Absent
- Type 2C: None or Few
- Internal nuclei: In older patients; Up to 25% of type 2 fibers
- Coexpression: Some muscle fibers express both fast & slow myosin
- No rods: Even with ultrastructure evaluation
- Type 1 muscle fibers
- Also see: Nemaline rod myopathy
- TPM3 Variant syndrome: Cap myopathy 1
176
- Epidemiology: 2 patients
- Genetics
- TPM3 Mutations: Arg168Cys; Glu237Lys
- Inheritance: Dominant or Sporadic
- Clinical
- Delayed motor milestones
- Kyphoscoliosis,
- Muscle atrophy: Generalized
- Ptosis
- Weakness
- Hypotonia
- Axial, Proximal & Distal
- Mild to Moderate
- Walking unsupported
- Respiratory
- Face
- Dysmorphic: Elongated face; High-arched palate; Retrognathia
- Laboratory
- Muscle pathology
- Caps: NADH+
- Type I fibers: Small
- Internal nuclei: Larger fibers
- Aggregates
- MRI: Masseter hypertrophy; Diffuse fatty replacement of muscle
- EMG: Myopathic
- Muscle pathology
- TPM3 variant syndrome: Congenital muscle stiffness, Hypercontractile
123
- Epidemiology: 2 patients (US & German)
- Genetics
- Mutations: Glutamic acid deletions; ΔE218, ΔE224
- Inheritance: de novo; Dominant
- Allelic disorders
- TPM3 protein: Mutation effects
- Muscle fibers: Maximal active tension reduced
- Ca++ sensitivity of troponin-tropomyosin complex: Increased
- Cross-bridge cycling kinetics: Abnormal
- Excitation-contraction coupling: Excessively sensitized
- Clinical
- Onset age: Congenital or Prenatal
- Fetal movements: Reduced
- Muscle
- Stiffness: Gait & Passive movement; Increased with heat
- Respiratory failure
- Joints: Reduced range of movement; Thoracic kyphosis; Spine rigidity; Short stature
- Muscle relaxants: No clear benefit
- Laboratory
- EMG: Myopathy + Denervation; No spontaneous activity
- Muscle ultrasound: Increased echogenicity
- Serum CK: 200 to 1,000
- Muscle pathology
- Fiber size: Moderately varied
- Internal architectgure: Mildy irregular
- Ultrastructure: Z-band streamning of mini-rods
- Congenital stiffness: Differential diagnosis
- CRYAB
- ACTA1
- TPM3
- Also see: Arthrogryposis
NEM2 Rod myopathy
● Nebulin (NEB)
- Epidemiology
- Most common form of recessive "Typical" nemaline rod myopathy
- Birth incidence: 1:50,000
- Genetics: Mutations
- > 380 mutations: 95% small variants
- Point mutations: Most common
- Missense variants: Common; Conserved actin- & tropomyosin-binding sites likely pathogenic
- Common mutations
- Ashkenazi Jews
- 2,502-bp deletion in exon 55 & parts of introns 54 and 56
- Carrier frequency: 1 in 108
- Severity: "Typical" forms of nemaline myopathy
- Chinese: c.21417+3A>G exon 144 splice
- Ashkenazi Jews
- Other mutation types
- Small insertions, deletions or point mutations
- Truncating (55%)
- Duplications (10% to 15%)
- Location: Exons 82-105; NEB TRI region
- Eight exons repeated 3 times in 32-kb triplicate
- Normal copy #: 6
- ≥ 2 extra: Nemaline myopathies
- 3' end of gene: Exons 165 to 185; Z-disc part of nebulin
- Mutations producing no nebulin protein
- Severe disease
- Death at 1 day to 9 months
- Splice site
- No inversions or translocations
- No clear relation between recessive mutation types & phenotype
- Dominant mutation
- Large deletions (52 to 97 exons)
- Results in expression of smaller nebulin protein
- Clinical: Distal NM in Finnish family
- Allelic disorders: Severe congenital, or Intermediate (mild) forms of Rod myopathy
- Rod myopathy
- Nebulin myopathy, Distal, Recessive
- Lethal multiple pterygium syndrome (Fetal akinesia; Arthrogryposis-6 (AMC6))
- Core-Rod myopathy
- Cap & Rod myopathy, Recessive
- Cap & Rod Distal myopathy, Dominant
- Nebulin mutations NOT identified in: Adult onset rod myopathy
- Nebulin (NEB) protein
- Missense mutations: May cause myofilament structural changes 165
- Clinical: Infantile onset; Mild to moderate severity ("Typical")
- Onset: Birth; Some movement present
- Weakness
- Trunk
- Distal legs: Ankle dorsiflexors
- Knee flexors
- Face
- Nasal voice
- Respiratory
- More severe forms
- Obstructive sleep apnea with dysmorphic face
- Tongue: Weak; Triple furrow
- Dysmorphic features
- HEENT
- High arched palate
- Micrognathia: May require surgical correction
- Contractures: Fingers
- Chest deformaties
- Joint hypermobility
- HEENT
- Cardiac: Normal
- Extraocular muscles: Normal
- Progression
- Slow or None
- May improve after initial respirator dependance
- Muscle pathology
- Nebulin: Still present
- Fiber size: Varied
- Fiber types
- Type 2 muscle fibers: Reduced numbers in milder disease
- 2X myosin: Absent in younger patients with more severe disease
- Co-expression of fast & slow myosin: Intermeditate disease severity
- Neonatal myosin: More severe disease
- Rods
- Ultrastructure
- Z disk aberrations
- Myofibrillar dissociation & smallness: Severe disease
- Contain: α-actinin; myotilin, telethonin, actin, tropomyosin & desmin
- Present in normal-sized & small muscle fibers
- Severe disease: Globular
- Milder disease: Elongated; Subsarcolemmal & Cytoplasmic; More common
- Ultrastructure
- Muscle MRI
53
- Mild phenotype
- Involvement: Tibialis anterior, Soleus
- Sparing: Thigh muscles
- Moderate phenotype involvement
- Rectus femoris, Vastus lateralis, Hamstring, Anterior leg, Soleus
- Mild phenotype
- Nebulin Variant: Core-Rod myopathy
80
- Epidemiology: 1 patient
- Genetics
- Recessive
- Nebulin Mutations
- Duplications
- Truncation
- g.195187_195188dupAC in exon 140: Loss of all nebulin isoforms
- g.234878_234881dupTCAA in exon 171: Loss of some nebulin isoforms
- Clinical
- Onset
- Age: Congenital
- Hypotonia
- Respiratory insufficiency
- Weakness
- Diffuse
- Axial predominance
- Mild facial
- No independent walking
- Skeletal: Scoliosis; Joint contractures
- Cardiac: Normal
- Onset
- Laboratory
- EMG: Myopathic
- NCV: Normal
- Muscle Pathology: Muscle fibers with
- Rods: also Z-line streaming
- Cores: Similar to RYR1 mutations
- MRI: Abnormal soleus & Anterior + Lateral compartments; Gastrocnemius spared
- Other Core-Rod myopathy: RYR1 mutations
- Nebulin Variant: Distal Nebulin myopathy, Recessive
- Nebulin Variant: Cap & Rod Distal myopathy, Dominant
164
- Epidemiology: 18 families
- Genetics
- Inheritance: Dominant, de novo, Mosaic
- Mutations: Deletions, Large
- Sizes
- 52 to 97 exons
- Larger than: Deletions in recessive syndromes
- Sizes
- In-frame
- May be heterozygous or mosaic
- South Africa: Exon 14�77 deletion
- Nebulin protein
- Mutation results in expression of smaller nebulin
- Clinical
- General: Mild phenotype
- Onset
- Age: 1 to 48 years; Median 3 to 14 years
- Foot drop
- Weakness: Distal+
- Neck: Flexors
- Elbow flexors
- Hands: Wrists & Fingers (Extensors weaker than Flexors)
- Legs: Tibialis anterior & Toe extensors
- Proximal legs: Mild weakness
- Asymmetry (40%)
- Course
- Slow progression
- Most remain ambulant
- Muscle atrophy: Distal; Arms & Legs
- Dysmorphism: High-arched palate; Narrow face
- Sensory loss (40%): Small fiber modalities; Distal
- Laboratory
- Muscle pathology
- Myopathic
- Type 1 fiber predominance
- Nemaline rods: Older patients
- Cap-like structures
- Core-like structures
- Serum CK: Normal
- MRI: T1 signal in anterior tibial
- Muscle pathology
- Epidemiology: 1 patient
- Genetics
- Inheritance: Recessive
- Nebulin mutations: c.1152+1G>A; c.1782+4_1782+5delAG
- Clinical
- Onset age: Infancy
- Hypotonia
- Weakness: Global; Fatigue; Wheelchair
- Ptosis: Bilateral
- Respiratory failure
- Swallowing difficulty
- Tendon reflexes: Reduced
- Developmental disability
- Morphology: Head
- Dolichocephaly
- Ears: Low-set; Attached lobes
- Upper lip: Tented
- Mouth: Downturned corners
- Palate: High arched
- Skeletal
- Clinodactyly
- Hyperconvex nails
- Toes: 1st & 2nd overlapping
- Cryptorchidism
- Laboratory
- NCV: Normal
- EMG: Normal
- Serum CK: Normal
- Muscle biopsy: 7 months
- Nemaline rods
- Fiber-type disproportion: Type 1 Small & Predominant
- Muscle biopsy: 6 years
- Cap-like structures
- Peripheral in muscle fibers
- Contain rods
- Toluidine blue positive
- Nemaline rods
- Ultrastructure: Z-disk & Myofibrillar abnormalities
- Cap-like structures
- Brain MRI: Normal
- Nosology: Arthrogryposis Multiplex Congenita-6 (AMC6)
- Epidemiology: 28 families
- Genetics
- Nebulin Mutations
- Types: Splice site, Deletion or Stop
- Inheritance: Recessive
- Nebulin Mutations
- Clinical
- Fetal akinesia
- Growth retardation: Intrauterine
- Limb contractures
- Pterygia
- Effusion: Pericardial; Hydrothorax
- Face: Hypertelorism; Downslanting palpebral fissures, Long philtrum, Low set ears
- Muscle
- Nemaline rods
- Fiber sizes: Varied
NEM3 Rod myopathy (CMYO2)
● α-Actin (ACTA1; Skeletal muscle)
- Nosology: NEM3; CMYP2; CMYO2
- Epidemiology
- 23% of rod myopathies
- > 50% of severe rod myopathies
- Genetics
- Actin (ACTA1) protein
- α-Skeletal muscle actin
- Normal adults: Present in muscle but not heart
- Development: Becomes predominant isoform in muscle at 3rd trimester of gestation
- Dominant syndromes: Mutant ACTA1 exerts dominant negative effect
- Aggregation may be caused by
- Heat shock
- Leptomycin B (a specific inhibitor of nuclear export mediated by leucine-rich nuclear export signals)
- Mutation in leucine-rich nuclear export signals
- α-Skeletal muscle actin
- Clinical
- Subtypes
- CMYO2A, Dominant
- CMYO2B, Recessive
- CMYO2C, Dominant, Severe, Infantile
- Scapuloperoneal (SHPM)
- Congenital muscular dystrophy + Rigid spine, Recessive
- Nemaline myopathy + Intranuclear rods
- Congenital myopathy + Excess of thin filaments
- Cap myopathy
- Myofibrillar myopathy
- Cytoplasmic body myopathy
- Inclusion body & Rod myopathy
- Distal myopathy
- Fiber type disproportion, Congenital
- Zebra body
- Cardiomyopathy: Dilated > Hypertrophic
- Arthrogryposis/Fetal akinesia
- Hypercontractility: Lys328Asn
- CMYO2A, Dominant
- Myopathies: Dominant inheritance (CMYO2A)
- Clinical: Varied phenotypes
- General
- Onset age
- Varied within families
- Severe: May be antenatal or neonatal
- Cardiac function: Normal
- Onset age
- Mild
- ACTA1 Mutations: Asn115Ser; Gly268Cys; Ile136Met
- Clinical
- Onset: Age 12
- Weakness: Trunk & Proximal; Mild facial
- Respiratory failure: 5th decade
- Progression: Slow over decades
- Laboratory
- Serum CK: Normal
- MRI: Gluteal & Anterolateral thigh involvement
- Muscle morphology
- Rods, especially in small fibers; Core-like structures
- Muscle fiber hypertrophy: Ile136Met
- Scapuloperoneal (SHPM)
120
- Genetics
- ACTA1 mutation: Glu197Asp
- Inheritance: Dominant
- Clinical
- Onset age: Infancy to 3rd decade
- Weakness: Scapuloperoneal & Distal
- Wrist extensor
- Finger drop
- Foot drop
- Quadriceps (50%)
- Scapular winging
- Face: Mild
- Contractures: Ankle; Elbow; Shoulder
- Tendon reflexes: Reduced or Absent
- Laboratory
- Muscle pathology
- Type I fiber atrophy
- Internal architecure irregular
- Internal nuclei
- Serum CK: Normal to Slightly high
- Muscle pathology
- Genetics
- Severe (CMYO2C)
85
- ACTA1 genetics
- New dominant mutations
- N94K; L144F; Arg183Gly; G270R; Ile357Leu
- Clinical
- Phenotypes
- Lethal; Congenital
- Congenital weakness
- CNS involvement: Some patients
- Developmental delay
- Frontal lobe hypoplasia
- Lateral ventricle dilation
- Other organs: Few patients
- Skeletal dysplasia
- Hepatomegaly
- Urinary tract stenosis
- Phenotypes
- Muscle pathology
- Fiber size: Varied; Small fibers in clusters
- Nemaline rods: Cytoplasmic & Intranuclear
- Cytoplasmic bodies (Asn94Lys mutation) 140
- Thin filament aggregates
- Endomysial fibrosis
- Asymmetric weakness
- Genetics: Mosaic (de novo) mutations (Ala116Pro; Asp156His) 167
- Clinical
- Onset: Neonatal or Infant
- Weakness: Asymmetric; Proximal & Distal; Respiratory
- Cardiomyopathy: 1 patient
- Nemaline myopathy with intranuclear rods
- Cap myopathy
- Other mutations: Met132Val; Val163Met; Met269Arg
- General
- Pathology
- Fiber types
- Abnormal differentiation
- Type I: Small
- Glycogen accumulation
- Rods
- No relation between abundance & disease severity
- More in small muscle fibers
- Stain for α-actinin 2
- Rods tend to be in type 1 muscle fibers
- May be present in some muscles but not others
- Some syndromes with no rods
- Fiber morphology
- Myofibrillar disruption
- Whorling of actin thin filaments
- Fiber types
- Clinical: Varied phenotypes
- ACTA1 variant: CMYO2B; Rod myopathy, Recessive inheritance
66
- ACTA1 Genetics
- Truncation mutations identified; ? Missense
- Truncation: Arg41X (French), Tyr364fsX (Spanish), Asp181fsX10 (British)
- Clinical
- Weakness
- Severe in most: Diffuse; Hypotonia
- Respiratory failure
- Feeding difficulties
- Death in 1st year: Most
- Contractures (30%)
- Course: Survival through childhood in some
- Weakness
- Laboratory
- Serum CK: Mildly elevated or normal
- Muscle
- Morphology: Nemaline rods & Zebra bodies
- Actin: Skeletal α-actin absent; Cardiac α-actin increased
- Lipid: Increased
- ACTA1 Genetics
- ACTA1 variant: Congenital muscular dystrophy with Rigid spine
128
- Epidemiology: 1 Sri Lankan family, 2 brothers
- Genetics
- Mutations: Homozygous; Missense; c.460G>C, p.Val154Leu
- Inheritance: Recessive
- Clinical
- Onset age: Infancy
- Weakness
- Neck
- Falling
- Distribution: Generalized
- Muscle size: Small
- Skeletal
- Rigid spine
- Kyphoscoliosis
- Laboratory
- Skeletal muscle
- Fiber size: Varied
- Internal nuclei
- Fiber splitting
- NADH: Central lucencies
- Skeletal muscle α-actin: Present
- Cardiac muscle α-actin: Increased
- Ultrastructure: Smeared & accumulated Z-band material; Rare rods
- Serum CK: 222 to 1900
- NCV: Normal
- Muscle MRI: Fatty repleacement
- More severe: Posterior & Rectus femoris
- Relative sparing: Vastus lateralis & medialis, Gracilis, Sartorius
- EKG: Normal
- Skeletal muscle
- ACTA1 variant: Nemaline myopathy with Intranuclear rods
62
- Genetics
- Rod myopathy Mutations: All involve ACTA1
- Inheritance
- Sporadic, de novo (Most) > Dominant
- Marked phenotypic variation in family
- Mutations: Most missense
- Clinical
- Onset age: Birth (Most) to 55 years
- Arthrogryposis
- Narrow face
- Thin musculature
- Hypotonia
- Weakness: Diffuse
- Respiratory failure
- Disease course: Early death in severe cases
- Laboratory
- Serum CK: Normal or Mildly elevated
- EMG: Myopathic
- Muscle
- Intranuclear rods
- Probably formed within nucleus
- Larger than sarcoplasmic rods: Up to length of several sarcomeres
- One or multiple per nucleus
- Frequency: 2% to 80% of nuclei
- Number of fibers with intranuclear rods correlates with disease severity
- Perinuclear space
- Enlarged
- Abnormal localization: Lamin A/C, Nesprin-1, Nesprin-2
- Cytoplasmic bodies
- Myofibrillar disorganization
- Endomysial connective tissue: Increased insome
- Cardiac α-actin: Increased in longer surviving cases
- Intranuclear rods
- Genetics
- ACTA1 variant: Cap myopathy with ACTA1 mutation
82
- Epidemiology: 1 patient
- Genetics
- Mutation: Met47Val
- Inheritance: Dominant
- Clinical
- Decreased fetal movements
- Birth: Poor respiration
- Motor
- Hypotonia: Axial & Peripheral
- Weakness: Severe
- Minimal spontaneous movements
- Generalized atrophy
- Deep tendon reflexes: Absent
- General
- Low hairline
- Micrognathia
- High arched palate
- Single palmar crease
- Long fingers
- Undescended testes
- Death: 5 years
- Laboratory
- Serum CK: Normal
- Head MRI: Normal
- Muscle biopsies
- 5 weeks: Type 1 smallness; No caps
- 4 years
- Caps: Contain thin filaments, α-Actinin, Actin & Desmin; NADH+
- Internal nuclei
- Expanded Z-bands
- ACTA1 variant: Myofibrillar myopathy, Congenital
121
- Epidemiology: 1 patient
- Genetics
- Inheritance: Sporadic, Dominant
- Mutation: In-frame 2-amino-acid insertion; c.437_442dupCCTCCG; p.146_147dupAlaSer
- Clinical
- Onset age: Congenital
- Hypotonia
- Motor delay
- Weakness
- Arms > Legs
- Face: Diplegia
- Tendon reflexes: Reduced
- Dysmorphic features
- Ears: Low-set
- Contractures: Metacarpophalangeal & proximal interphalangeal joints involving most fingers
- High arched palate
- Death: 3 years
- Laboratory
- Brain MRI: Normal
- Serum CK: Normal
- EMG: Fibrillations; Motor units small
- Muscle biopsy
- Fiber morphology: Varied size; Splitting; Internal nuclei
- Fiber architecture: Vacuoles; Myofibrillar disorganization; Hyline structures
- Fiber aggregates: Desmin; αB-crystallin, Myotilin, Dystrophin, NCAM
- Fiber damage: Necrosis & Regeneration
- Endomysial connective tissue: Increased
- Perimysium: Widened
- Differential diagnosis: Childhood-onset myofibrillar myopathies
- BAG3
- CRYAB, Recessive
- ACTA1: Congenital; Child
- PYROXD1
- ACTA1 variant: Myofibrillar myopathy, Child onset
166
- Epidemiology: Italian family, 6 patients
- Genetics
- Inheritance: Dominant
- Mutation: c.148G>A; p.Gly50Ser
- Clinical
- Onset: Infant or Childhood
- Face: Weak; Temporal atrophy
- Weakness
- Neck
- Diffuse: Proximal > Distal
- Course: Slow progression
- Laboratory
- Serum CK: Usually normal
- Muscle MRI
- Abnormal: SCM, Rectus abdominis, Gluteus minimus, Vastus intermedius, Gastrocnemius
- Symmetric
- Muscle pathology
- Fiber sizes: Varied
- Internal nuclei
- Nemaline rods
- Cytoplasmic aggregates: Dark on gomori trichrome; Contain Desmin, Myotilin & α-B crystallin
- ACTA1 variant: Cytoplasmic body myopathy, Congenital
140
- Epidemiology: 3 patients
- Genetics
- Inheritance: Sporadic; Dominant
- Mutation: Asn94Lys; Heterozygous
- ACTA1 protein
- Clinical
- Onset age: Congenital
- Hypotonia
- Weakness: Severe; Respiratory
- Laboratory
- Muscle: Cytoplasmic bodies
- ACTA1 variant: Inclusion body & Rod myopathy
149
- Epidemiology: 1 family, 4 patients
- Genetics
- Inheritance: Dominant
- Mutation: Gly50Asp
- Allelic disorders
- Clinical
- Onset age: Childhood
- Hypotonia: Axial
- Weakness
- Face
- Trunk
- Neck flexor
- Abdominal
- Distal
- Fingers: Flexor ± Extensor
- Toe dorsiflexors
- Proximal
- Legs: Quadriceps; Hip flexors
- Arms: Shoulders; elbows
- Slow running
- Course: Slow progression
- Other: Scoliosis & Scapular winging in 1 patient
- Laboratory
- Muscle
- Vacuoles, rimmed
- Rods: On ultrastructure
- Type 1 fibers: Small
- Serum CK: Mildly high
- Muscle
- ACTA1 variant: Distal myopathy
150
- Epidemiology: 4 families
- Genetics
- Inheritance: Dominant
- Mutations
- Gly253Arg: Finger flexor & anterior leg weakness
- Gly50Asp: Finger flexor weakness
- Glu197Asp: Scapuloperoneal ± Rods
- Clinical
- Onset age: Childhood to Teen-age
- Weakness
- Distal: Anterior distal leg; Finger extensor
- Proximal: Hips; Later in life
- Trunk (Older patient): Respiratory; Camptocormia
- Course: Slow progression in adulthood
- Other: Pes cavus; Joint hyperlaxity
- Laboratory
- Muscle
- Atrophic muscle fibers
- Rods
- Myofibrillar pathology (Actin or Myotilin aggregates): 1 patient
- May have rimmed vacuoles
- Serum CK: Normal
- EMG: Mixed neurogenic & myopathic
- Muscle
- Subtypes
- Muscle MRI
- Involved muscle signal: Increased T1
- Involved muscles: Diffuse involvement of thigh and leg
- Relative sparing: Gastrocnemius
NEM4 Rod myopathy (CMYO23)
● β-Tropomyosin (TPM2)
- Nosology: NEM4 Rod myopathy; CMYP23; CMYO23
- Epidemiology: Unusual cause of rod myopathy
- Genetics
- TPM2 mutations: 29 identified; Lys7del, E117K, Glu139del, Q147P
- TPM2 allelic disorders: Most dominant
- Distal arthrogryposis 1A: Dominant or de novo
- DA2B4: Dominant
- Cap myopathy 2: Dominant
- Escobar syndrome: Recessive
- Congenital fiber type disproportion 5: Dominant
- Nemaline rod myopathy (NEM4): Dominant or Recessive
- β-Tropomyosin (TPM2) protein
190
- Mutation abnormalities
- General: Dominant effect on regulation of actin�myosin interactions
- Altered interactions
- Affinity for actin
- Tropomyosin head-to-tail binding
- Ca++ activation of TPM contractility
- Increased Ca++ sensitivity: Hypercontractile molecular phenotype
- More limb (93%) or jaw (30%) contractures
- More associated with: Arthrogryposis
- Non-hypercontractile mutations: Often hypocontractile
- Axial contractures more common (90%)
- More associated with: Myopathies; Nemaline, Rod-Core, Cap, Fiber disproportion
- Increased Ca++ sensitivity: Hypercontractile molecular phenotype
- Mutation abnormalities
- Clinical
- Onset: Infancy to Childhood
- Weakness
- Onset age: Adolescent or Adult
- Proximal > Distal
- Some with respiratory failure
- Less severe than TPM3
- Course: Slow progression; Some lose independent ambulation in adulthood
- Contractures: Variable
- Common: Distal & Proximal joints; Arms & Legs
- Some patients: Few or None
- Cardiac: Normal
- Laboratory
- Serum CK: High
- Muscle MRI
- Muscle involvement: Masticatory; Distal leg
- Muscle biopsy
- Nemaline rods: Often subsarcolemmal
- Fiber size: Varied
- Type 1 fibers: Predominant; Small
- Core-like regions: Some patients
- TPM2 Variant: Escobar syndrome
74
- Epidemiology: Consanguenous Algerian family
- Genetics
- Inheritance: Recessive
- Mutation
- Homozygous
- Gln210Stop: Exon 6b, specific for skeletal muscle β-TM isoform 1
- Allelic disorders
- TPM2 protein: Absent skeletal muscle isoform of β-tropomyosin
- Clinical
- Onset: Congenital
- Motor: Hypotonia; Delayed milestones
- Respiratory distress
- Joints: Pterygia; Arthrogryposis
- Cardiac: Bifascicular ventricular block with right bundle block & left anterior hemiblock
- Muscle
- Connective tissue: Abundant
- Rods: More common in type I muscle fibers (60%)
- TPM2 Variant: Congenital Fiber Type Disproportion 5
95
- Genetics
- Inheritance: Dominant
- TPM2 Mutations: Missense; Ser61Pro, Ala155Val
- Allelic disorders
- Clinical: Variable severity
- Neonatal; Well or Hypotonic
- Motor milestones: Delayed
- Weakness
- Proximal
- Face
- Respiratory in more severe patient
- Mild to Moderate
- Laboratory
- Muscle pathology
- Type 1 fiber diameter: Small
- Type 2 fiber diameter > Type 1: 50% to 60% difference
- Ultrastructure: Small caps in 1 patient
- Serum CK: Normal
- Muscle pathology
- Genetics
NEM5A: Rod myopathy, Severe Infantile (Amish type)
● Troponin T1 (Skeletal, Slow; TNNT1)
- Epidemiology: 18 families
- Genetics
- Mutations
- Stop
- Older Order Amish: Nonsense; E180X; Homozygous
- Hispanic patient: c323C>G (S108X)
- Palestinian: Complex truncating indel mutation
- Other: Spice site; Exon 14 deletion
- Missense
- French-Canadian: c.287T>C (p.Leu96Pro)
- Later onset: Arg184Pro
- Stop
- Allelic disorders
- Nemaline myopathy, Dominant (NEM5C)
- Mutations: Missense; Asp65Ala, E104V
- Onset: Infancy & Childhood
- Weakness: Moderate; Proximal
- Course: Slow progression
- Congenital weakness, severe, Recessive
188
- Mutations: Nonsense
- Onset: Infancy
- Weakness: Hypotonia; Respiratory
- Course: Early death
- Nemaline myopathy + Rigid Spine & Respiratory Insufficiency (NEM5B)
- Nemaline myopathy, Dominant (NEM5C)
- Mutations
- TNNT1 protein
- Link between troponin complex & tropomyosin (Tm)
- Helps to regulate muscle contraction
- Clinical
- Onset
- Usual: 1st months of life
- Less common: Adult
- Tremors
- Begin within few days of birth
- Distribution: Jaw & Lower limbs
- Resolve by 2 to 3 months
- Skeletal
- Contractures: Shoulders, Hips & Knees; Mild
- Thoracic dystrophy: Pectus carinatum: Rigid chest wall or spine; Proximal contractures
- Weakness
- Early
- Hypotonia
- Motor milestones: Delayed
- Proximal > Distal
- Respiratory
- Feeding difficulties
- Early
- Rhabdomyolysis: French-Canadian; Infection trigger; Dantrolene treatment
- Cardiac: No 1° involvement; Terminal right congestive failure
- Course
- Proximal contractures: Increase
- Many: Death from respiratory insufficiency < 2 years
- French-Canadian: May live & remain ambulatory to adulthood
- Adult onset: Progressive with respiratory failure
- Onset
- Laboratory
- Serum CK: Normal to 4x High
- Leg MRI
- Posterior thigh, Tibialis anterior & Deep posterior leg involvement
- Symmetric
- EMG: Mild myopathy or normal
- Muscle Pathology
- Type 1 fiber disproportion
- Z-band streaming
- Rods: Centrally placed
- Myofibrillar disruption
- Myofiber degeneration
- French-Canadian: Multi-minicores in type 1 fibers
NEM6 Rod myopathy with Slow movements
● Kelch-repeat and BTB (POZ) domain containing 13 (KBTBD13)
- Epidemiology: 32 European & Australian patients
- Genetics
- Mutations: Missense; Arg248Ser, Lys390Asn, Arg408Cys
- KBTBD13 protein
- Tissues: Skeletal & Cardiac muscle; Lung
- Subcellular: Cytoplasm; Sarcomere
- Interacts with: Cullin 3 ubiquitin ligase; Actin
- Required for formation of functional Cul3 RING ubiquitin ligase complex
- Involved in ubiquitination of proteins destined for degradation
- Mutation
- Actin binding: Altered
- Thin filament stiffness: Increased
- Ca++ dynamics: Altered
- Muscle contractile force: Reduced
- Relaxation: Slow
- Other BTB/Kelch protein disorders
- Clinical
- Onset age: Childhood
- Weakness
- Mild to Moderate
- Proximal & Neck flexors > Distal
- No respiratory insufficiency
- Muscle size: Atrophy in some patients
- Movements: Slow; Unable to run; Poor rapid postural correction
- Progression: Slow; Disability after 50 years
- Skeletal (Some patients): High Palate; Thorax deformities
- Cardiac: Normal
- Laboratory
- Serum CK: Normal
- EMG: Myopathic
- Muscle CT: Fatty infiltration of proximal limb & truncal muscles
- Muscle biopsy
- Type 1: Predominance; Hypertrophy
- Type 2: Atrophy
- Internal nuclei
- Rods: Granular; Diffusely distributed; All fibers
- Core-like structures
- Less sharply defined than typical cores
- Ultrastructure: Large regions of rods with loss of muscle structure
NEM7 Rod myopathy
● Cofilin-2 (CFL2)
- Epidemiology: 3 families
- Genetics
- Mutations: Missense (Ala35Thr, Val7Met); 4 base pair deletion
- Heterozygotes: Not affected
- Cofilin-2 protein
- Member of AC (ADF/Cofilin) protein group
- Regulate actin filament dynamics: Actin depolymerizing factors
- Other group members: Cofilin-1
;
Destrin
- Skeletal muscle specific isoform
- Localized to thin filaments
- Exerts effect on actin, in part, through interactions with tropomyosins
- Binds G- and F-actin in 1:1 ratio
- Major component of intranuclear & cytoplasmic actin rods
- Member of AC (ADF/Cofilin) protein group
- Clinical
- Onset: Hypotonia at birth
- Early motor milestones: Delayed
- Ambulation: Present for short distances; Frequent falls; Inability to run
- No facial weakness or foot drop
- Muscle pathology
NEM8 Nemaline Rod myopathy, severe
● Kelch-like family member 40 (KLHL40; KBTBD5; Sarcosynapsin; SYRP)
- Epidemiology: 28 kindreds
- European & Asian
- Most common severe nemaline myopathy in Japan (28%)
- Mutations
- Loss of function
- Homozygous or Compound heterozygous
- Types: Frameshift; Missense; Nonsense; Splice site
- Locations: All exons
- Mutation associations
- Arg500Cys: Mild phenotype
- Thr506Pro: Hong Kong Ethnic Chinese; Severe phenotype
- Glu528Lys mutation: Japanese & Kurdish; Slightly milder phenotype
- Kelch repeat disorders
- KHLH40 protein
- Clinical
- Variation in severity
- Onset: in utero
- Fetal akinesia or Hypokinesia
- Polyhydramnios
- Skeletal
- Arthrogryposis & Contractures (89%)
- Fractures (53%)
- Dysmorphic face, mild (100%)
- Chest deformity
- Motor: At birth
- Respiratory failure (97%)
- Swallowing difficulties (96%)
- Face weakness (100%)
- Ophthalmoplegia (17%)
- Limbs: Severe; Some with no antigravity movement
- Treatment
125
- Pyridostigmine (30 mg, 3x/day = 20 mg/kg/day)
- Ephedrine
- Death: Mean 5 months; Range 20 days to alive at 11 years
- Laboratory
- Muscle
- Rods are miliary
- Many
- In most muscle fibers
- Small: 200 to 500 nm in diameters
- Shape: Cuboidal
- Some only seen by EM
- Muscle fiber size: Varied
- Type 2 predominance: Some patients
- Endomysial connective tissue: Normal or Increased
- KLHL40 protein: Absent or Reduced
- Rods are miliary
- Hyponatremia
- Serum CK: Normal after neonatal period
- Muscle
NEM9: Nemaline Rod myopathy 9
● Kelch-like family member 41 (KLHL41; KBTBD10)
- Epidemiology: 4 families
- Genetics
- Mutations: Small deletions; Missense
- Clinical correlations
- Frameshift: Severe phenotype with early death; Arthrogryposis
- Missense: Weakness; Survival to late childhood or Early adult
- Allelic disorder
- KLHL41 protein in muscle
- Perinuclear
- Sarcomere: Near terminal cisternae over I-band
- Endoplasmic reticulum
- BTB-Kelch domain containing
- Interacts with: Nebulin, N-RAP (Nebulin-related anchoring protein), Actin
- Promotes assembly of myofibrils
- Mutations: Reduced levels of KLHL41 in muscle
- Clinical: Severe phenotype
- Fetal akinesia sequence
- Hypotonia
- Dislocation of hips & knees
- Facial dysmorphism: Micrognathia; Cleft palate
- Death: Cardiorespiratory arrest; Age 1 day
- Clinical: Intermediate phenotype
- Weakness
- Initial walking: 2 to 3 years
- Respiratory insufficiency: 5 years
- Dysarthria
- Wheelchair: 16 years
- Skeletal
- Palate: High arched
- Scoliosis
- Weakness
- Clinical: Mild phenotype
- Weakness: Distal > Proximal
- Contractures: Distal
- Muscle pathology
- Nemaline rods: No correlation with disease severity
NEM: Nemaline Rod myopathy 10 (NEM10)
● Leiomodin-3 (LMOD3)
- Epidemiology: 18 families
- Genetics
- Mutations: Mostly truncating; G326R; Gln335Arg; Leu550Phe
- Missense mutations: Milder course
- Germany & Austria founder mutation: Leu550Phe
- LMOD3 protein
- Cytoskeleton
- Tropomodulin family
- Skeletal > Cardiac muscle
- M line/thin filament pointed end region
- Actin nucleator
- Tropomyosin binding domain
- Binding partners: Nebulin & KLHL40
- Also see: LMOD2 CMD 2G
- Clinical: Severe, often lethal
- Onset age: Congenital (90%)
- Antenatal
- Polyhydramnios (62%)
- Fetal movements: Reduced (48%)
- Joint contractures, multiple (48%)
- Birth
- Premature delivery (35%)
- Hypotonia
- Weakness: General & Bulbar
- Respiratory insufficiency
- Feeding difficulties
- Ophthalmoplegia (29%)
- Face
- Weak
- Elongated
- Bulbar
- Dysphagia: May resolve with increasing age
- Jaw: Chewing difficulty
- Speech: Dysarthric
- Limbs: Weak
- Scoliosis: some surviving patients
- Course
- Severe type: Neonatal death in most
- Milder course
- Most ambulatory
- Patients survive to adulthood
- Laboratory
- Muscle MRI
- Sparing: Vastus lateralis, Gracilis, Semimembranosus, Semitendinosus, Extensor digitorum longus
- Muscle biopsy
- Nemaline rods: Atypical
- Present in smaller muscle fibers
- Clustered
- "Caterpillar"
- Fringed
- Endomysial connective tissue: Increased
- Sarcomeres: Often reduced or absent
- LMOD3 often absent
- Ultrastructure
- Nemaline bodies
- Short thickened Z-discs in doublets interconnected by filaments
- Surrounded by: Short, thin filament fringe
- Z-band streaming
- Myofilaments: Disorganized; Core-like structures
- Nemaline bodies
- Nemaline rods: Atypical
- Muscle MRI
NEM 11: Rod myopathy (CMYO24)
● Myopalladin (MYPN)
- Nosology: NEM11 Rod myopathy; CMYP24; CMYO24
- Epidemiology: 5 patients
- Genetics
- Mutations: Truncation & Splice site
- Allelic disorders
- MYPN protein
- Sarcomere component
- Location: Z- & I-bands
- Tethers nebulin
 in skeletal muscle &
nebulette
in cardiac muscle to α-actinin
(ACTN2
) at Z lines
in skeletal muscle &
nebulette
in cardiac muscle to α-actinin
(ACTN2
) at Z lines
- ACTN2 binding site: C-terminal & Central regions of MYPN
- MYPN-α-Actinin-Nebulin
- Can form a link tethering actin thin filaments & titin filaments in Z-line
- Assembles I-Z-I bodies in striated muscle
- N-terminal region: Binds to cardiac ankyrin repeat protein (CARP; ANKRD1]
; Attached to titin at I-band
- Nucleus
- In skeletal & cardiac muscle
- MYPN colocalizes with CARP
- Homologous protein: Palladin
- Sarcomere component
- Clinical
- Onset age: 4 to 25 years
- Weakness
- Legs ≥ Arms
- Distal & Proximal
- Neck
- Respiratory
- Progression: Slow
- Cardiomyopathy (50%): Hypertrophy; Hypokinesia
- Skeletal: Mild changes; Face Δ or High arched palate; No contractures
- Intellect: Normal
- Laboratory
- Heterozygous carriers: No cardiac disease
- MYPN variant syndrome: Congenital myopathy with Caps ± Rods
139
- Epidemiology: French, Asian & African patients
- Genetics
- Mutations: Truncation & splice site
- Allelic disorders
- Clinical
- Onset age: Congenital or Early childhood
- Weakness
- Diffuse: Axial; Limbs, Proximal & Distal
- Face: May be asymmetric
- Respiratory: Some patients
- Cardiac: Normal
- Laboratory
- Serum CK: Normal or Mildly high
- EMG: Myopathic
- Muscle MRI: Diffuse smallness; Glutei, Posterior thigh, Anterior leg
- Muscle biopsy
- Caps: Stain for Trichrome, PAS, NADH-TR, Myopalladin, Desmin, α-Actinin
- Rod-like structures: Atypical, Large, Rosette shape
- Type 1 muscle fiber predominance
CMYO20: Nemaline Rod myopathy
● Ryanodine Receptor 3 (RYR3)
- Nosology: CMYP20; CMYO20; Nemaline rod myopathy
- Epidemiology: 3 patients
- Genetics
- Mutations: Missense (Asp667Gly, Met2070Val, Arg2980Leu); Splice
- Disease relation: Uncertain 202
- RYR3 protein
- Location: Brain & Muscle
- Muscle
- Similar in both fiber types
- Striated
- Between double RYR1 rows
- Functions
- Mediates release of Ca++: From sarcoplasmic reticulum → cytoplasm
- Maintains Ca++ release during prolonged periods of Ca++ influx
- Mediates Ca++-induced Ca++ release from endoplasmic reticulum in non-muscle cells
- Activity regulated by calmodulin
- Clinical
- Onset age: Congenital to 5 years
- Weakness
- Face
- Proximal
- Symmetric
- Legs & Arms
- Vital capacity: 75%
- Tongue: Small
- Tendon reflexes: Reduced
- Skeletal
- Face: Long; Narrow
- Micrognathia
- Scapular winging
- Arthrogryposis
- Laboratory
- NCV: Normal
- RNS: Normal
- EMG: Myopathic; No spontaneous activity
- Serum CK: Normal
- Muscle pathology
- Fiber sizes: Varied; Type 1 small; Type 2 large
- Fiber types: Type1 predominance
- Rods: Perinuclear; Cytoplasmic; Subsarcolemmal
- Core-like areas in regions with nemaline bodies
NEM: Cardiomyopathy + Rods, Infantile 174
● Cyclase-associated actin cytoskeleton regulatory protein 2 (CAP2)
- Epidemiology: 1 patient
- Genetics
- Mutation: Homozygous; c.1288 delT (C430fs)
- Allelic disorders
- Dilated cardiomyopathy 2I
- 6q22 microdeletion
- CAP2 protein
- Clinical
- Onset: Birth
- Cardiomyopathy: Progressive; Dilated
- Skin: Soft; Wound healing abnormal
- Joints: Hyperlaxity
- Hypotonia
- Weakness: Face; Neck flexors; Ptosis (Lower lid)
- Tendon reflexes: Reduced
- Short stature
- Course: Maintenance with cardiac treatments
- Laboratory
- Pathology
- Rods: In skeletal & cardiac muscle
- Muscle: Fiber sizes varied; Type I predominance
- Echocardiogram: Decreased biventricular systolioc function
- Pathology
Rod myopathy: Mild 64
● Dominant
- Epidemiology: Swiss family
- Onset
- Age: Neonatal
- Hypotonia
- Clinical: Mild Course
- Walking: Onset 18 to 24 months
- Weakness: Axial & Proximal
- Difficulty running
- Most with no limitation in normal ADLs
- Muscle biopsy
- Type 1 muscle fiber predominance
- Rods: 1% to 5% of muscle fibers; Increase with age
Rod myopathy: Other forms
- Infantile onset types: Severe
- Fetal akinesia sequence
- Onset: 2nd trimester of pregnancy
- Polyhydramnion; Joint contractures (Multiple); Lung hypoplasia
- Not linked to α-actin, nebulin or α-tropomyosin 3
- Birth
- Diffuse Hypotonia, Weakness & Little spontaneous activity
- Respiratory failure
- Cardiomyopathy: Dilated
- Arthrogryposis
- Survival: Weeks to months
- Pathology: Cytoplasmic & intranuclear rods
- May need oil immersion lens to visualize
- Fetal akinesia sequence
- Rod myopathy, Adult onset (SLONM): Heterogeneous
59
- Nosology: Sporadic, Late-Onset Nemaline Myopathy
- Epidemiology
- Male = Female, but Male > Female with M-protein
- Disease associations
- HIV
- Monoclonal gammopathy
- Some patients with neither condition
- Genetics
- Family history: Often none
- Gene testing: Usually negative
- Clinical
- Onset
- Age
- 3rd to 9th decade: Mean 52 to 54 years
- Younger with HIV: Mean onset 4th decade
- Much older than inherited nemaline rod myopathies
- Subacute weakness: 4 to 8 weeks to nadir
- Age
- Weakness
- Limbs (85%)
- Proximal > Distal
- Arms & Legs
- Asymmetry (30% to 71%)
- Axial
- Posterior neck (50%)
- Camptocormia
- Cranial
- Face (25% to 60%)
- Dysphagia (50%)
- Dysarthria (30% to 50%)
- Respiratory
- Frequency: 50% to 100%; Reduced maximal respiratory pressure
- Occasionally presenting feature
- May be associated with M-protein
- Failure: 30%
- Severity: Common cause of death
- Face: Mild or None
- Limbs (85%)
- Pain
- Myalgia (60%)
- Neck or Back
- Cardiomyopathy: Ventricular; Uncommon
- Progression
- High mortality in 5% to 40%: Death within 1 year
- Slow progression over years in others
- M-protein: More weakness & progression
- 10 year survival: 68%
- Treatment
- Prednisone: Most response in HIV patients
- IvIg
- Chemotherapy ± Autologous stem cell transplantation
- Worse outcome
- Long disease course before treatment
- No reduction in M-protein levels
- Onset
- Laboratory
- Muscle biopsy
208
- Muscle Fiber sizes: Varied; Small fibers round or angular
- Rods
- Frequency
- Median: 11%
- Range: 4% to 40% of muscle fibers
- Present in fewer fibers than inherited rod myopathies
- Molecular
- May be best seen on trichrome in 3 μM sections
- Distribution: Diffuse in muscle fiber cytoplasm
- May
- Only occur later in disease course
- Be intranuclear
- Frequency
- Rod-bearing muscle fibers may also have
- Smaller size
- Large vesicular nuclei
- Focal cytoplasmic basophilia
- Small vacuoles
- Loss of ATPase activity
- NCAM stain
- Immunoglobulin overexpression: In Rod-containg areas; κ-light chain
- MHC-I expression in cytoplasm
- Fiber type: I or II
- Cytoplasmic bodies
- Mitochondrial pathology: COX- fibers common
- Inflammation
- Frequency (30%): CD3 & CD8 cells; Mild when present
- Location: Endomysial or Perivascular
- Necrotic &amo regenerating muscle fibers: 50% to 70%
- Lobulated muscle fibers (50%)
- Type I fiber predominance (20%)
- MHC I expression by muscle fibers (75%): Diffuse or Multifocal
- Endomysial connective tissue: Moderately increased
- Ultrastructure: Rods < 1μM in length
- Downregulated proteins vs inherited rods: DPYSL3, MYH7, NRAP
- M-protein
- Frequency: 50%
- Type: IgG κ or λ: 50% to 80%
- Prognosis: No long term effect or ? Unfavorable outcome
- Serum CK: Usually normal; 65 to 240
- EMG
- Irritable myopathy: Fibrillations in > 80%
- Motor unit potentials: Mixed short & long duration
- Muscle MRI
- Trunk: Neck extensors; Paraspinous; Subscapularis
- Legs: Gluteus medius & minimus (50%), Hamstring (Semimembranosus (60%) & Biceps), Soleus (45%)
- Asymmetric (73%)
- Muscle biopsy
208
- Rule out
- Sarcoglycanopathies
- HIV nemaline rod myopathy
- Hereditary nemaline rod myopathy
- IM-VAMP
- Treatment: Uncertain; ? Prednisone
- Central core disease, Rods & Malignant Hyperthermia
Centronuclear (Myotubular) Myopathy
89
|
Types Dominant CNM1: Dynamin 2; 19p13 CNM modifier: MTMR14 ? CNM3: MYF6; 12q21 CNM4: CCDC78; 16p13 CNM: BIN1: 2q14 Recessive CNM2: BIN1: 2q14 CNM5 (+ DCM): SPEG; 2q35 CNM6: ZAK (MAP3K20): 2q31 CNM: RYR1: 19q13 CNM: TTN; 2q31 CNM: NOTCH2; 1p12 CNM: GLDN; 15q21 Canine (Lab): PTPLA Canine (Lab & Springer): EHBP1L1 X-linked CNMX: MTM1; Xq28 Carriers Mouse CNM in type 2 fibers: SRPK3-null Differential diagnosis Myotonic dystrophy 1, Congenital Pathology |
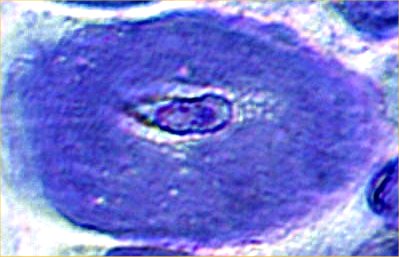
|
- Centronuclear myopathies: Differential Clinical features
119
- Onset age
- Clinical features
- Pathology
- Also see: Congenital Weakness DDx
- Centronuclear myopathies: Common Molecular mechanisms
191
- Endocytic proteins involved in T-tubule biogenesis
- Membrane remodeling
- CNM protein functions
- Centronuclear myopathy: X-linked (CNMX; MTM1; XLMTM)
● Myotubularin (MTM1) ;
Chromosome Xq28; Recessive
;
Chromosome Xq28; Recessive
Genetics
MTM1 protein
Clinical
Laboratory
Female carriers
PIP Phosphatase disorders
- Epidemiology: 1 male in 50,000
- Gene features
- 15 exons: Translation start codon in exon 2
- Gene expression
- Ubiquitous
- Smaller transcript selectively expressed in skeletal muscle & testis
- Has characteristics of housekeeping function
- High GC content of MTM1 promoter
- No defined TATA-box or CAAT-box structure
- Gene Mutations
- General
- > 530 different disease related mutations identified 10
- Mutation Locations
- Widely distributed throughout gene: All 15 exons involved
- Spread unevenly across gene
- Hotspots: 6 recurrent mutations
- Codon 48 frameshift; P205L; R224X; R241C; 420insFIQ; R421X
- Arg241Cys: 2nd most frequent mutation; Causes mild (72%) or severe myopathy
- Mutations in Males > Females
- Mutation types
- 60% null variants
- Point mutations: Missense (55) & Nonsense (40); Most between exons 8 & 12
- Small insertions or deletions (50)
- Large deletions (15)
- Splice site mutations (38)
- IVS11, A-G, -10: Most common mutation; Causes severe myopathy
- Germline mosaicism identified in some families
- Mutation locations
- Most frequent in exons: 4 > 12 > 3 > 8 > 9 > 11
- Highly conserved regions
- ? Reflects importance of mutated gene regions to functional domains
- Genotype-Phenotype Correlations
199
- Truncating mutations
- 95% of truncating variants associated with severe phenotype
- Frequent death or ventilator dependent
- Muscle fiber size: Smaller
- Recurrent MTM1 mutations: Arg37X; Lys47del
- Some mild phenotypes with very distal mutations
- 95% of truncating variants associated with severe phenotype
- Missense & Single amino acid deletion mutations
- Higher prevalence of mild phenotypes (33% to 45%)
- Individual missense mutations may cause mild or severe phenotypes
- Recurrent MTM1 mutations: Arg69Cys; Pro205Leu; Arg241Cys; Arg421Gln
- All in phosphatase active site or SID domain have severe phenotype
- Protein alteration
- Predicted severe: 99% with severe phenotype
- Normal level of myotubularin: Missense mutation or Small deletion or insertion
- Large chromosomal deletions: Associated abnormal genital development
- Some symptomatic female carriers
- Truncating mutations
- Many mutations can be explained by
- Methylation-mediated DNA repair of modified CpG dinucleotides, or
- DNA polymerase slippage with short tandem-repeated sequences
- Disease modifier: Overexpression of ubiquitous isoform of DNM2 related to more severe disease
- Allelic disorders
- X-linked centronuclear myopathy (XLCNM)
- Myotubular myopathy: Animals
- Dogs
- Labrador retriever (N155K)
- Rottweiler (Q384P)
- Boykin Spaniel (Arg512X)
- Maine coon cat (Ala152Val)
- Dogs
- Myotubular myopathy + Abnormal genital development
- Contiguous gene syndrome
- Xq27.3-q28 duplication syndrome
- General
- MTM1 protein
- Type: PIP phosphatase
- Tissue localization: Ubiquitously expressed
- Cell localization
- Sarcolemma
- I band, including triads
- Associated with endosomes
- ? Nuclear
- MTM1 structure
- Contains active site of protein tyrosine phosphatases (PTP)
- Similarity to: Dual-specificity serine/tyrosine phosphatases (dsPTPases)
- MTM1 functions
- Enzyme action: Dephosphorylates phosphatidylinositol 3-phosphate
- Associates with SET (Suvar3-9, Enhancer-of-zeste, Trithorax) domains
- SET domains: In proteins contributing to epigenetic mechanisms of gene regulation
- Roles
- Muscle fiber maturation
- Plasma membrane homeostasis
- Muscle: Formation & maintenance of "triad"
- Overexpression
- Induces the formation of filopodia-like projections
- Affects protein trafficking from late endosome to lysosome
- Muscle
- Proliferation of membrane structures that contain myotubularin
- Vacuoles labelled by markers of sarcolemma & T-tubules (caveolin-3, dystrophin, DHPR)
- MTM1 interacts with: SPEG; Desmin
- Myotubularin family & related disorders
- PIP phosphatase disorders
- Clinical
- Onset
- Infancy
- Respiratory failure
- Pregnancy
- Polyhydramnios: 50%
- Fetal movement: Decreased
- Premature delivery 31%
- Skeletal: Large head (70%); Narrow face (80%); Long digits (60%)
- Motor
- Severe Hypotonia: At birth
- Weakness: Proximal & Distal; Symmetric
- Respiratory Insufficiency: 90% need external support
- Ophthalmoplegia & Ptosis
- Onset age: At birth (60%), or with disease progression
- Prognosis
- Early death: Mortality 44%
- Mean = 5 to 10 months
- 64% < 18 months
- ? Improvement after neonatal period
- Weakness course: Non-progressive
- Survivors often
- Respirator dependent (80%)
- Feeding tube dependent
- Require head support
- Never ambulate
- Early death: Mortality 44%
- Systemic features in some survivors > 1 year
- GI
- Pyloric stenosis
- Gallstones
- Hepatic: Peliosis hepatis; Cholestasis
- Hematologic
- Spherocytosis
- Bleeding diathesis: Vitamin K-responsive
- Renal: Stones; Calcinosis
- Skeletal: Rapid linear growth; Advanced bone age
- Cognitive function: Reduced
- GI
- Treatment: Some benefit from pyridostigmine or albuterol
- Onset
- Laboratory
- CK: Normal or mildly elevated
- EMG
- Motor unit potentials: Small amplitude; Polyphasic
- Spontaneous activity: Fibrillations; Positive sharp waves; Complex repetitive discharges
- Neuromuscular Junction testing: May be abnormal
86
- Stimulated single fiber EMG: Abnormal in 1 patient
- Muscle pathology
67
- Single central nucleus
- In some muscle fibers: Range = 4% to 40%
- Frequency may increase with age
- Present in chains on longitudinal sections
- May occur in both fiber types
- Perinuclear: Vacuole-like areas
- Type 1 muscle fiber predominance
- Muscle fiber size
- Average fiber size: Reduced
- Increase in size with age: Reduced
- Type 1 muscle fibers smaller than type 2: More prominent in older patients
- Fiber size smaller in more severe disease
- Fiber size smaller with truncation/deletion mutations than missense mutations
- Central areas of fibers
- Reduced myofibrillar ATPase reaction
- Aggregations of organelles: Mitochondria, Lysosomes, SR
- Increased oxidative enzyme activity & glycogen staining
- Periphery of fibers
- Oxidative enzyme activity: Pale halo
- Single central nucleus
- Myotubularin mutation: Carriers
182
- Frequency of manifesting carriers: 50%; 10% with severe disease
- Mild syndrome
- Genetics: Missense mutations
- Clinical
- Weakness: Face, Mild
- Skeletal: Asymmetry
- More severe
56
- Genetics
- Mutations: Stop; R253X; c.1354�1G>A (Splice site); Large deletion
- Skewed X-inactivation
- Several families
- Minor relation to disease
- Other mechanisms: Reduced expression of other allele
- Incomplete penetrance
- Clinical
- Onset
- 1st decade
- Arm weakness
- Gait disorder
- Skeletal asymmetry
- Weakness
- Early: Arm
- Proximal
- Distal: With disease progression
- Respiratory
- Chest wall
- Hemidiaphragm: Elevated
- FVC: 23% to 100%
- Asymmetry
- Progressive
- In 3rd to 5th decades
- May lose ambulation
- Respiratory
- Skeletal
- Asymmetry: Face; Smaller hand on side with more weakness
- Kyphoscoliosis
- Pes equinovarus (Bilateral)
- Onset
- Laboratory
- Serum CK: Normal
- Muscle pathology
- Varied fiber size: Both fiber types
- Internal nuclei
- NADH: Normal internal architecture or Necklace fibers
- Muscle MRI: Abnormal in weak muscles; Asymmetric
- Genetics
- Neonatal onset
39
- Genetics
- Mutation: 605delT
- Non-familial case
- Clinical
- Hypotonia
- Joint laxity
- Waddling gait
- Weakness: Limb-girdle; Facial
- Tendon reflexes: Absent
- Genetics
- Myotubularin knock-out mouse
37
- Progressive weakness
- Muscle fiber development
- Initially normal with peripheral nuclei
- Later: Degenerative changes & central nuclei
- Pathology of muscle fibers
- Variable among muscles: Most in biceps
- In type I & II muscle fibers
- Atrophy
- Mis-localized mitochondria & nuclei
- Serum CK: Mildly elevated
- Tissues other than muscle: Spared
- Centronuclear Myopathy: Autosomal Recessive
- General
- Onset
- Infancy, Childhood, Adult < 30 years: ~1/3 each
- Possible subgroups
49
- Early onset with ophthalmoplegia
- Early onset without ophthalmoplegia
- Late onset without ophthalmoplegia
- Clinical
- Weakness: Proximal > Distal
- Ophthalmoplegia: Early onset patients
- Facial weakness
- Usually survive past infancy
- Onset
- General
- Centronuclear myopathy 2 (CNM2)
● Bridging integrator 1 (BIN1; Amphiphysin 2);
Chromosome 2q14.3; Recessive
68 or Dominant
- Epidemiology: 7 families
- Genetics
- Mutations
- Types: Missense, Splice & Stop; K35N, D151N, K575X
- Effect: Partial loss of function
- Homozygous: Common
- Allelic disorders
- Centronuclear myopathy, Dominant, Mild, Adult-onset
- Great Dane canine myopathy
- Mutations
- BIN1 protein
- Involved in membrane remodeling
- Regulated by: Phosphoinositides
- Role in organization of T tubules
- Mutations
- Loss of functional link to Dynamin 2
- ? Disordered T tubule biogenesis
- Malignant cells: Often reduced
- Clinical: Severe CNM
- Onset
- Age: Birth to Childhood (8 yrs (D151N mutation))
- Pregnancy: Reduced or normal fetal movements
- Weakness
- Proximal
- Mild
- Ophthalmoparesis & Ptosis: Some patients
- Bulbar: Face; Dysarthria
- No respiratory failure
- Muscle atrophy: Diffuse
- Skeletal
- Facial dysmorphism, Mild
- Contractures: Birth
- Cardiac: Dilated in some patients
- Course
- Slowly progressive weakness
- Survival through childhood: 60%
- Onset
- Laboratory
- Serum CK: Variably high
- Electrodiagnostic
- Repetitive stimulation: Decrement
- EMG: Myotonia & Pseudomyotonia; Myopathic units
- Pathology
- Fiber sizes: Less variability than other CNM types
- Fiber types
- Homogeneous population of rounded hypotrophic type I fibers
- Type I predominance
- Central nuclei
- In many fibers
- Clusters & Chains: Several nuclei in central part of fibers
- Shapes: May be abnormal
- NADH
- Central nuclei (clear region) surrounded by rim of densely stained material
- Radial organization of sarcoplasmic reticulum: Some patients
- Endomysial connective tissue: Increased
- Necrosis & Regeneration: Not present
- Caveolin 3: Immunoreactivity in centrally located vacuoles in some fibers
- T tubule marker dihydropyridine receptor-a (DHPRa): Immunoreactivity around centrally located nuclei
- BIN1 variant syndrome: Centronuclear myopathy, Dominant, Adult onset
116
- Epidemiology: 5 European families
- Genetics
- Inheritance: Dominant or Sporadic
- Mutations
- Types: Missense; Inframe deletions; Single base pair deletions in stop codon causing larger protein
- c.1778delC, c.1779delA, c.1780delT, Arg24Cys, Lys21del
- BIN1 protein
- Clinical
- Onset age: 22 to 50 years
- Weakness
- Proximal > Distal
- Legs > Arms
- Face & Respiration: Normal
- Gait disorder or Wheelchair: Some patients
- Progressive
- Eyes: Ptosis (25%); Vertical gaze palsy (25%)
- Cognitive: Normal
- Cardiac: Normal
- Laboratory
- Muscle
- Type I fibers: Predominant (65% to 92%) & Hypotrophy
- Nuclei: Central & Clustered
- Muscle fiber central staining: Dynamin 2, Caveolin-3, p62
- Sarcoplasmic strands, Radial
- Serum CK: Normal to High x10
- MRI muscle
- Distal leg: All muscles, especially posterior
- Proximal: Patchy muscles; 1 patient with sartorius & adductor longus
- EMG: Myopathic
- Muscle
- Centronuclear myopathy (CNM3): Autosomal Dominant
● MYF6;
Chromosome 12q21.31; Dominant
- Epidemiology: 1 patient
- Genetics
- MYF6 gene mutation: Ala112Ser
- Ala112Ser: Also occurs at low frequency in random databases
- MYF6 mutation in Becker dystrophy: Converts disease to more severe phenotype
- MYF6 protein
- Muscle determination factor
- Helix--loop--helix domain: Common feature of myogenic factors
- Clinical
- Onset: Late Childhood - Adult
- Weakness: Proximal > Distal
- Leg cramps
- ± Ophthalmoplegia, Facial, or Scapular weakness
- Laboratory
- CK: Normal or Mildly elevated
- Muscle biopsy: Myopathic; Ring fibers; Central nuclei
- Centronuclear myopathy: Autosomal Dominant (CNM1)
60
● Dynamin 2 (DNM2);
Chromosome 19p13.2; Dominant or Sporadic
- Epidemiology
- Multiple centronuclear myopathy families
- Common in Netherlands
- Genetics
- Mutations
- Types: Misense
- Locations
- Exons 8 & 11
- Middle domain
- Involved in self assembly & centrosomal location of protein
- Mild or intermediate phenotype
- Pleckstrin homology domain: Severe phenotype
- Effects: Gain of function
- de novo: Common
- Clinical-Genetic correlations
- Severe disease: Phe372Ser, Ser619Leu, Ala618Asp, Glu368Lys
- Later onset: Arg522His; Arg369Trp
- Common variant with varied presentation: Arg465Trp
- CNM mutations: Most located in autoinhibitory interface
- CMT mutations: Most located in lipid binding loops of PH domain
- Allelic disorders
- CMT, Dominant-intermediate Type B (CMTDIB): Mutations in plecstrin domain
- CMT 2M
- Centronuclear myopathy (CNM1)
- Centronuclear myopathy, Severe neonatal with good prognosis
- Centronuclear myopathy with cataracts
- Lethal congenital contracture syndrome 5 (LCCS5): Homozygous mutation
- Hereditary spastic paraplegia, Adult onset 181
- MTMR14
: Possible modifier gene
- Dnm2R465W/+ mouse
168
- Satellite cells (Pax7+): Reduced
- Muscle regeneration: Impaired
- Mutations
- DNM2 protein
191
- Expression: Ubiquitous & Muscle specific isoforms
- Associated with microtubules
- Family: Large GTPases
- Binds to BIN1 & SNX9
- Implicated in
- Endocytosis & Cell motility
- Regulates tubular invagination of plasma membrane
- Part of cellular fusion-fission apparatus
- Membrane: Remodeling & Trafficking processes
- DNM2 oligomers form: Helical structure around neck of Nascent vesicle
- GTP hydrolysis: Leads to vesicle release in cytoplasm
- Endocytosis & Cell motility
- DNM2 increased with MTM1 mutations
- DNM2 mutations
- Abnormal organelle positioning in myofibers
- Nucleus centralization
- Sarcoplasmic reticulum & mitochondria mis-position
- Overexpression of ubiquitous isoform: More severe MTM1 CNM disease
- Gain-of-function: GTPase & Membrane fission activities high
- Abnormal organelle positioning in myofibers
- Clinical
- Onset
- Age: Variable
- Usual
- Ages: Neonatal or Childhood (80%)
- Early milestones: Delayed motor or Normal
- Earlier onset
- Some severe, dominant or sporadic patients
- Some patients: Adolescence & Adult
- Usual
- Symptoms
- Neonatal: Hypotonia; Respiratory insufficiency
- Exercise-related muscle pain
- Weakness
- Difficulty walking & climbing stairs
- Falls
- Poor at sports
- Age: Variable
- Weakness
- Distal > Proximal: Upper extremity all distal
- Axial: Neck flexors; Abdomen
- Ocular: Ptosis; Ophthalmoplegia (50%); Strabismus (50%)
- Face 40%
- Respiratory: Common with neonatal onset; Restrictive
- Bulbar: Dysphagia (40%); Difficult chewing or swallowing
- Gait: Often (90%) limited
- Progression: Slow; Some patients in wheelchair in 6th decade
- Fatigue: May respond to pyridostigmine
- Muscle size
- Atrophy: Distal (30%)
- Hypertrophy: Paravertebral
- Tendon reflexes: Often absent or reduced
- Contractures
- Ankles (90%)
- Fingers
- Jaw: Limited motility (30%)
- Skeletal
- Palate: High arched (47%)
- Spine (70%): Scoliosis or Hyperlordosis
- Foot: Pes cavus
- Other systemic changes in a few patients
- Retinopathy
- Neutropenia
- Cardiomyopathy: In female carrier
- Onset
- Laboratory
- Serum CK: Normal, usually; Mildly high, rarely
- NCV: Reduced CMAP; NCV some mildly slow
- EMG: Myopathic; Some with spontaneous activity or pseudomyotonia
- Repetitive stimulation: Decrement or Normal
- MRI: Early involvement of distal, posterior leg
- Involved: Lateral soleus, Gastrocnemius; Posterior thigh; Adductor longus; Biceps; Semimembranosus
- Spared: Sartorius, Gracilis & Rectus femoris
- Muscle
- Nuclei
- Central: Some in chains
- Type 1 muscle fibers: Predominant; Small
- NADH: "Spoke-like" (radial) strands (40%)
- Central region of fibers in regions without nuclei stains for: PAS, phosphorylase & Oxidative enzymes
- Core-like lesions
- In more preserved proximal muscles
- Stain for: Desmin; DHPRα1s; RYR1
- Multi-minicores (20%)
- Necklace fibers (30%)
- Necrosis: Not present
- Ultrastructure
- Central nuclei: Often surounded by mitochondria & other organelles
- Central spaces without nuclei have: Mitochondria, Sarcoplasmic reticulum, Golgi complex & Glycogen particles
- Radial distribution of intermyofibrillar sarcoplasmic strands
- Nuclei
- Variant: Severe CNM, Neonatal onset with good prognosis
71
- Family history: Sporadic or Dominant
- Genetics: DNM2 mutations
- De novo, Dominant
- Heterozygous
- Exon 16: Plekstrin Homology domain, N- & C-terminal regions
- Types: Missense or In frame
- Ala618Thr; A618D; Ser619Leu; Ser619Trp; V625del
- Onset
- Neonatal
- Hypotonia
- Clinical
- Pregnancy: Normal
- Motor
- Hypotonia; Weak suck
- Cranial nerve: Face weakness; Ptosis; Ophthalmoparesis; Strabismus
- Weakness: Generalized; Distal > Proximal; Legs > Arms
- Respiratory weakness: Restrictive syndrome common
- Age at walking: Usually normal
- Jaw contractures
- Skeletal: Pes cavus
- Cardiac function: Normal
- Peripheral nerve: Normal
- Prognosis
- Good
- Slow improvement to mid-childhood or adult
- Some patients in wheelchair in 2nd to 6th decade
- Laboratory
- Serum CK: Normal to Slightly increased
- EMG: Myopathic; Some with spontaneous activity or pseudomyotonia
- NCV: Occasional axonal neuropathy
- Muscle
- Type I fibers: Predominance & Smallness
- Nuclei
- Central (3% to 50%): In both fiber types; Some in chains
- Perinuclear clearing
- Large fibers: May be both types
- Radial sarcoplasmic strands: Some
- Endomysial connective tissue: Variably increased
- Variant: CNM with Cataracts
- Genetics
- DNM2 mutation: Leu621Pro
- Allelic disorders
- Clinical
- Onset: Neonatal
- Hypotonia
- Respiratory insufficiency
- Cataracts
- External ophthalmoplegia
- Course: Progressive; Death at 14 years
- Genetics
- DNM2 variant: Lethal congenital contracture syndrome 5 (LCCS5), Recessive
97
- Epidemiology: 3 Pakistani infants in 1 family
- Genetics
- DNM2 mutation: Phe379Val; Homozygous
- Allelic disorders
- Clinical
- Akinesia
- Joint contractures
- Hypotonia
- Skeletal abnormalities
- Hemorrhages: Brain & Retinal
- Laboratory
- Muscle: Central nuclei
- Epidemiology
- Centronuclear myopathy, Recessive
110
● Titin (TTN);
Chromosome 2q31.2; Recessive
- Epidemiology: 5 patients
- Genetics
- Mutations: Truncating or Splice; Compound Heterozygous
- Allelic disorders
- Clinical
- Onset age: Congenital to 3 years
- Weakness
- Distribution: Diffuse; Proximal + Distal
- Face
- Respiratory
- Ambulation: Present in 60%
- Feeding difficulty: Occasional
- Eye movements: Normal
- Tendon reflexes: Usually absent
- Scoliosis (80%)
- Cardiac: Normal or Dilated cardiomyopathy
- Cognition: Normal
- Laboratory
- Serum CK: Usually normal; May be up to 1,400
- EMG: Often myopathic
- Muscle biopsy
- Nuclei
- Central in 5% to 25% of muscle fibers
- Internal, not central: 20% to 70% of muscle fibers
- Multiple internal in single fiber: Often
- NADH Internal architecture: Central clear (core-like) regions in some muscle fibers
- Type 1 fibers: Predominance (60% to 80%); Smallness
- Fiber size: Variation
- Endomysial connective tissue: Increased in some patients
- Titin C-terminal & Calpain-3 staining: Reduced in some patients
- Ultrastructure: Myofibrillar & sarcomere disorganization in fibers with internal nuclei
- Nuclei
- Centronuclear myopathy 5 (+ Dilated cardiomyopathy, Recessive) (CNM5)
113
● Striated muscle preferentially expressed protein kinase (SPEG; APEG1)
;
Chromosome 2q35; Recessive
- Epidemiology: 13 patients
- Genetics
- Mutations: 15 identified; Stop (Gln2233*, Arg1426*, c.3709_3715�29del36, c.3709_3715�29del36); Missense (Gly2757Val; Thr2904Ala)
- SPEG protein
- Clinical
- Onset age: Birth
- Hypotonia
- Weakness: Longer surviving patients
- Motor milestones: Delayed
- Proximal
- Face
- Feeding difficulty: G-tube needed
- Respiratory insufficiency
- Ophthalmoplegia
- Tendon reflexes: Reduced
- Skeletal
- Palate: High arched
- Contractures: Hip
- Arthrogryposis
- Circulatory
- Cardiomyopathy, dilated
- Pulmonary Arterial Hypertension
- Course: Death at 3 weeks to Surviving at 6 years
- Laboratory
- Serum CK: 22 to 280
- Muscle biopsy
- Central nuclei
- Fiber size: Small
- Fiber type: I predominant
- SPEG staining: Reduced
- Centronuclear myopathy
203
● NOTCH2;
Chromosome 1p12; Recessive
- Epidemiology: 1 family, 2 patients
- Genetics
- Mutation: Ile1689Phe; Homozygous
- Allelic disorders
- Alagille syndrome 2, Dominant
- Hajdu-Cheney syndrome, Dominant
- Alagille syndrome 2, Dominant
- NOTCH2 protein
- Clinical
- Onset age: 2 to 3 months
- Hypotonia
- Weakness: Diffuse; Proximal > Distal
- Skeletal: Scoliosis; Ankle contractures
- Laboratory
- Muscle pathology
- Type 1 fibers: Small
- Central or internal nuclei: Type 1 fibers
- Subsarcolemmal aggregates
- Muscle pathology
- Centronuclear myopathy: Autosomal Dominant
● Centronuclear myopathy: Genetically undefined phenotypes- Clinical subtype 1: Typical Dominant centronuclear myopathy
49
- Epidemiology: 2 families
- Onset age: Childhood to Adult
- Clinical
- Weakness
- Severity: Mild
- Distribution: Diffuse; Proximal & Distal; Occasional distal predominant
- Progression: Slow
- Disability: Mild; Most ambulatory to at least 6th decade
- Ptosis: Most
- Contractures: Occasional patient
- Tremor: Postural; Some patients
- Weakness
- Laboratory
- Serum CK: Normal
- EKG: Normal
- Pulmonary function: Normal or Reduced
- EMG: Myogenic
- Clinical subtype 2: Typical Dominant centronuclear myopathy with muscle hypertrophy
49
- Epidemiology: 1 family
- Onset: Adolescence & Adult, Rare infant
- Clinical
- Muscle hypertrophy: Diffuse
- Weakness
- Slowly progressive
- Distal arms
- Ocular
- Ptosis: Common
- Ophthalmoplegia: Occasional patients
- Skeletal
- Rigid spine: Mild 40%
- Ankle contractures: 30%
- Finger contractures: 14%
- Laboratory
- Serum CK: High (2x to 4x) in some patients
- EMG: Myopathic
- FVC: May be reduced
- Clinical subtype 1: Typical Dominant centronuclear myopathy
49
- Centronuclear myopathy: Muscle Biopsy
- Central nuclei
- Central pallor on ATPase
- Type 1 fibers predominant & small (50%)
- NADH: Radial distribution of sarcoplasmic strands
- 50% of carriers have central nuclei in muscle fibers
- Endomysial fibrosis: More severe recessive cases
- Centronuclear myopathy: Canine
50
● Protein tyrosine phosphatase-like (Proline instead of catalytic arginine), Member A; (PTPLA; HACD1)); 10p12.33 (Canine chromosome 2); Recessive
- Epidemiology: Labrador retrievers
- Carrier frequency: 15%
- Common in many parts of world
- Genetics
- PTPLA mutation
- Insertion: g.9459-9460ins238
- Founder: Arose 50 years ago
- Allelic with: Human CFTD
- K64Q polymorphism: Found in arrhythmogenic right ventricular dysplasia but not likely pathogenic
- PTPLA mutation
- PTPLA (HACD1) protein
- Nosology: 3-hydroxyacyl-CoA dehydratase 1 (HACD1)
- Mainly expressed in skeletal muscles & heart
- Subcellular: Endoplasmic reticulum resident enzyme
- Catalyzes 3rd reaction of elongation of very long chain fatty acids (VLCFA)
- Role in myoblast growth & differentiation
- Clinical
- Onset
- Age: 2 weeks
- Weight loss
- Muscle
- Atrophy: Proximal legs; Cervical; Temporal
- Hypotonia
- Neck: Ventroflexion (Head drop)
- Posture: Abnormal
- Gait: "Bunny hopping"
- Tendon reflexes: Absent
- Mega-esophagus
- Variable severity
- Course
- Progressive over 1st year
- Then stable
- Life span: Normal
- Onset
- Laboratory
- Muscle pathology
- Varied fiber size: Many small fibers
- Central nuclei: In larger & some smaller muscle fibers
- Type 1 muscle fibers: Predominance; Small
- EMG: Fibrillations
- Serum CK: Normal
- CNS: Normal
- Muscle pathology
- CMYO11: Human HACD1 (PTPLA) disease; Congenital fiber size disproportion (Type 1 muscle fibers small & prodominant)
109
- Nosology: MYONP; CMYO11; CMYP11
- Epidemiology: 3 families
- Genetics
- Mutations
- Type: Trp153Stop; Tyr248Stop; c.739_740delins1250; c.373_375+2delGAGGT; c.785-1G>T
- Homozygous
- Allelic with: Canine centronuclear myopathy
- Mutations
- HACD1 (PTPLA) protein
- Clinical
- Onset: Congenital; Hypotonia
- Weakness
- Face: Drooling; Reduced suck
- Head lag
- Feeding problems
- Weak cry
- Apnea: Recurrent (40%)
- Motor delay: Late walking
- Hypotonia: Especially proximal
- Proximal: Gowers sign; Waddling gait
- Course: Gradual improvement
- Tendon reflexes: Reduced
- Cognition: Normal
- Skeletal: Pes cavus; Pectus excavatum
- Laboratory
- Muscle biopsy
- Type 1 muscle fibers: Small; Predominant (80%)
- Type 2 muscle fibers: Occasional hypertrophy
- Internal nuclei: Few fibers
- No necrosis, cores or rods
- Dystrophy proteins: Normal
- Serum CK: Normal
- EMG: Normal or Myopathic
- Muscle biopsy
- Epidemiology: Labrador retrievers
Congenital Myopathy: Cardiac Arrhythmia ± Muscle weakness (CARDAR)
● Triadin (TRDN)
- Nosology
- Ventricular tachycardia, catecholaminergic polymorphic, 5 ± Muscle weakness (CPVT5)
- Triadin knockout syndrome
- Ventricular tachycardia, catecholaminergic polymorphic, 5 ± Muscle weakness (CPVT5)
- Epidemiology: > 20 patients
- Genetics
- Mutations: Null, Splice, Missense
- Allelic disorder: Long-QT Syndrome & Pediatric Sudden Cardiac Arrest
- Triadin protein
- Expression: Cardiac & Skeletal muscle
- More abundant in type 2 muscle fibers
- Muscle: Terminal cisternae
- Associated with
- RYR1: Present in cores
- Functions
- Clinical
- Weakness
- Onset age: Congenital or in utero
- Early: Hypoactive or Hypomotile
- General functions
- Fatigue
- Walk short distances
- Strenuous activities: Reduced
- Muscles
- Upper extremities: Suprascapular, Pectoralis major, Deltoid, Finger extensors, Interossei
- Trunk: Abdominals
- Legs: Iliopsoas, Anterior tibial, Toe extensors
- Face & Eyes: Normal
- Tendon reflexes: Reduced
- Cardiac
- in utero: Bradycardia
- Ventricular fibrillation
- ? Long QT syndrome
- Exercise-induced cardiac arrest in early childhood
- Weakness
- Laboratory
- NCV, EMG & SF-EMG: Normal
- RNS: Normal
- Serum CK: Normal
- EKG
- T-wave inversion in precordial leads V1�V4
- QT prolongation: Consistent or Transient
- Muscle biopsy
- Gomori trichrome: Abnormal spaces in muscle fibers with dot-like purple membranous material
- Triadin staining: Reduced
- Ultrastructure
- Lateral cisterns of sarcoplasmic reticulum: Focal dilation
- Triadin anchors from preserved lateral cisterns: Loss or Degeneration
- Triadin variant syndrome: Long-QT Syndrome & Pediatric Sudden Cardiac Arrest
28
- Epidemiology: 5 families
- Genetics
- Mutations: Frameshift; Homozygous or Compound heterozygous
- Allelic disorders
- Triadin protein
- Clinical
- Onset age: Childhood
- Cardiac arrest: Exercise induced
- Laboratory
- Stress test: Ventricular ectopy
- ECG: T-wave inversions in precordial leads V1 through V4
CMYO15: Congenital Myopathy + Neonatal Respiratory Insufficiency
● Troponin C, Fast (TNNC2)
- Nosology: MYONRI; CMYP15; CMYO15
- Epidemiology: 2 families, 4 patients
- Genetics
- Mutations: Missense; Asp34Tyr, Met79Ile
- TNNC2 protein
- Clinical
- Onset: Neonatal or Prenatal
- Prenatal: Polyhydramnios; Fetal movements reduced
- Weakness
- Respiratory insufficiency: Stridor
- Feeding difficulties
- Vocal cord paralysis
- Face
- Eyes: Ptosis; External ophthalmoplegia
- Gait: Waddling
- Skeletal
- Scoliosis
- Joint contractures: Jaw; Distal
- CNS: Learning difficulties, mild
- Course: Slow improvement
- Laboratory
- Muscle pathology: Fiber sizes varied; Type 2 smallness
- Muscle MRI: Focal fat infiltration
Congenital Muscular Dystrophy (MDC)
Congenital Muscular Dystrophy: Typical Features
- Inheritance: Autosomal recessive (AR)
- Frequency: Common cause of AR neuromuscular disorders
- Genetics: Causes
- Clinical features
- Onset age: Congenital or Infancy (< 1 year)
- Weakness: Diffuse
- Contractures
- CNS
- Common in severe forms of CMD
- May be subclinical
- Disorders of myelin or neuronal migration
- Muscle Pathology
- Myopathic
- Muscle fibers
- Size: Varied; Small & Some large
- Necrosis: Unusual after 4 years of age
- Endomysial connective tissue: Increased in more severe disorders
- Muscle fibers
- Disorders of connective tissue molecules
- Laminin α2: MDC1A
- Collagen
- Disorders of glycosylation
48
- Marker molecule: α-Dystroglycan
- Common O-mannosyl glycan structure: NeuAcα2-3Galα1-4GlcNAcα1-2Man-Ser/Thr
- Disorders: Defective O-mannosylation of α-dystroglycan
- Specific disorders
- Nosology
- MDDGA: Brain, Eye & Muscle involvement (Walker-Warburg)
- MDDGB: Congenital muscular dystrophy-dystroglycanopathy + Impaired intellectual development
- MDDGC: Muscular dystrophy-like syndromes
- Nuclear membranes: Lamin AC
- Other
- Myopathic
Fukuyama congenital muscular dystrophy (MDDGA4): Muscle-Eye-Brain
205
●
Fukutin (FKTN)
- Nosology: Muscular Dystrophy-Dystroglycanopathy (Congenital with brain and eye anomalies), Type A, 4
- Epidemiology
- Japan: Common (2.9 per 100,000); 1,000 to 2,000 patients; Incidence is 40% of Duchenne MD
- Western countries: Rare
- Genetics
- Mutations
- 80% of Japanese patients have similar haplotype
- Retrotransposal
insertion (3kb) of tandemly repeated sequences (Ancestral genotype)
- Location: 3' untranslated end of fukutin gene
- Effect of mutation: Reduced fukutin transcription
- Up to 100% of Japanese patients at least heterozygous for tandem-repeat insertion (Ancestral genotype)
- Retrotransposal
insertion (3kb) of tandemly repeated sequences (Ancestral genotype)
- Other mutations
- Japanese patients: Stop codons; Point mutations & small deletions
- Turkish patient: Homozygous 1 bp Insertion (nt504(insT)) in exon 5
- Ashkenazi Jews
76
- Mutation: c.1167insA; Homozygous
- Carriers: 0.7% of American Ashkenazi Jews
- Phenotype: Severe
- 80% of Japanese patients have similar haplotype
- Genetic-Clinical Correlations
- Milder phenotype: Homozygous for retrotransposon
- Severe phenotype: Heterozygous for retrotransposon & point mutation
- More ocular involvement
- Homozygosity for point mutations
- WWS-like syndrome reported
- May be lethal
- Allelic disorders
- Congenital muscular dystrophy (Muscle-Eye-Brain; MDDGA4)
- Congenital muscular dystrophy (No mental retardation; MDDGB4)
- LGMD 2M (MDDGC4)
- Cardiomyopathy, Dilated 1X (CMD 1X)
- Mutations
- Fukutin protein
17
- Brain mRNA
- Expressed in similar levels in adult & fetal brain
- Adult: Cortical neurons
- Fetal: Neuronal precursor & Migrating cells
- Cellular location: Neurons; Not astrocytes
- Protein
- Expressed in Golgi apparatus
- ? More abundant in fetal brain
- Possibly secreted
- Skeletal muscle: Not detectable by immunocytochemistry
- Function
- α-Dystroglycan glycosylation
- Catalyzes addition of ribitol to carbohydrate moiety
- Transfers ribitol-phosphates from cytidine diphosphate
- Intercellular interactions between retinal blasts & Müller cells
- Brain mRNA
- Clinical
- General
- Brain: Milder involvement than Walker-Warburg or Muscle-Eye-Brain
- Eye: Only occasionally severely affected
- Clinical heterogeneity: Some patients walk & have longer survival
- Clinical features: Severe cases
- Motor deficits
- Onset: Months to 1 year
- Hypotonia: Severe
- Weakness & Wasting: Face & Limbs
- Spasticity: Occasional
- Face & Bulbar
- Diplegia
- Large cheeks
- Dysphagia: May require tracheostomy
- Skeletal
- Contractures
- Kyphoscoliosis
- Skull: Microcephaly; Asymmetry
- CNS
- Seizures (50%)
- Mental retardation: Severe; IQ 30 to 50
- Hydrocephalus: Progressive
- Ocular
- Myopia
- Retinal hypoplasia & detachment
- Strabismus
- Persistent primary vitreous body & hyaloid artery
- Microphthalmos
- Optic nerve atrophy
- Blindness: From anterior & posterior chamber eye malformations
- Cardiomyopathy
- Onset age: 10 to 15 years
- Left ventricular
- Progressive
- Course
- Progressive disability
- Death: Often < 11 years; Respiratory dysfunction
- Motor deficits
- Clinical features: Milder or typical cases
- Typical peak motor function: Unassisted sitting
- Mildest cases: Walk unassisted
- General
- Laboratory
- CNS pathology
- Cortex
- Migrational defects
- Altered radial & tangential neuronal migration
- Micropolygyria (Type II lissencephaly), cobblestone
- Absence of lamination in gray matter
- Breaks in glia limitans & basal lamina
- Fukutin mRNA in Fukuyama CMD brain
- Absent in regions of overmigrated dysplasia
- Moderate expression in more normal areas
- Migrational defects
- White matter
- Hypomyelination
- Absent corpus callosum & septum pellucidum: Some patients
- CT scan: Lucencies, especially Frontal, Occipital regions spared
- Posterior fossa
- Brainstem: Kinking
- Pons: Flattening
- Cerebellum: Hypoplasia
- Posterior encephalocele
- Older patients
- Cerebellar efferent pathway: Dentate nucleus, Superior cerebellar peduncle & Red nucleus
- Lateral thalamic nucleus
- Cortex
- Serum CK: Moderately high
- EMG: Myopathic
- Muscle pathology: Myopathic
- Connective tissue: Increased
- Muscle fiber size: Variable
- Internal nuclei
- Differential involvement of muscle fascicles
- Basal lamina: Disrupted
- Muscle proteins
- α-dystroglycan: Greatly reduced in surface membrane & NMJs
- Laminin α2: Reduced
- CNS pathology
- Variant syndromes
- Walker-Warburg (MDDG4A)
- LGMD 2M
- Cardiomyopathy (CMD 1X)
- Walker-Warburg (MDDG4A)
Congenital muscular dystrophy: Merosin
(laminin α2-chain) deficient (MDC1A)
169
●
Laminin α2 (LAMA2; α2β1γ1; Lm211)
|
|
Normal Merosin: "Pure" form of congenital MD● Recessive
|
|
||||
Congenital MD with Integrin α-7 Deficiency
●
Integrin α-7 (ITGA7)
- Epidemiology: 3 Japanese patients
- Genetics
- Mutation: 98 base pair deletion most common
- Allelic disorder
- Cardiac disorder, Adult onset 195
- α7β1 Integrin protein
- Subunit tissue specificity
- α7: Muscle specific
- β1: Expressed in many tissues
- Abundance: α7β1 integrin is primary integrin in muscle
- Functions
- Laminin receptor
- Transmembrane link: Cytoskeleton to Extracellular matrix
- Myoblast migration
- Formation of
- Myotendinous junctions
- Postsynaptic membrane
- Changes in other myopathies
- Increased in Duchenne & Becker MD
- Reduced in Fukuyama & Laminin α2 deficient congenital MD
- Subunit tissue specificity
- Clinical
- Motor
- Hypotonia
- Delayed milestones
- Walk: Some patients; At 2 to 3 years
- Proximal weakness
- Respiratory involvement: With disease progression
- Mental retardation in 1 patient
- Motor
- Lab
- CK mildly elevated
- MRI: Normal brain
- Pathology, Muscle
- Fiber size variation
- Reduced staining for integrin α-7
Congenital MD with Joint Hyperlaxity 63
● ? Integrin-α9 (ITGA9)- Epidemiology: French-Canadian families from Southwestern Quebec
- Clinical: Similar to, but milder than, Ullrich CMD
- Onset
- Age: Birth
- Hypotonia with Contractures
- Weakness
- Generalized
- Walking (100%): 1 to 3 years
- Progression
- Slow
- Wheelchair in 2nd to 4th decade
- Respiratory
- Vital capacity: Reduced; Mean 50%; Range 21% to 100%
- No progression
- No failure
- Joints & Skeletal
- Contractures: Ankle (71%), Knee (21%), Shoulder (21%)
- Hyperlaxity
- Distal + Other
- Fingers (93%), Wrists (43%), Toes (43%), Elbows (43%), Cervical spine
- Scoliosis: Variable; Mild to Severe
- No protruding calcani
- Intelligence: Normal
- Onset
- Laboratory
- Serum CK: Normal to 1000
- Cardiac: Normal
- Muscle Pathology
- Fiber size: Variation
- Small fibers: Rounded or Angular
- Hypertrophy
- Central nuclei
- Endomysial connective tissue: Increased
- Type I muscle fiber predominance
- Rimmed vacuoles: In scattered muscle fibers
- Normal: α-dystroglycan; Collagen VI
- Fiber size: Variation
- Heterozygotes: Joint hyperlaxity
Congenital MD with CNS atrophy & Absent large myelinated peripheral nerve axons
●
Recessive
- Clinical
- Onset: Neonatal
- Arthrogryposis multiplex congenita
- Psychomotor retardation: Severe and generalized
- Motor
- Hypotonia
- Weakness: Face; Axial & limb
- Dysmorphic features
- Course: Respiratory failure & Death in 1st year in some
- Pathology
- Ventriculomegaly
- Atrophy: Severe of cerebrum and cerebellum
Congenital MD with cerebellar atrophy
●
? Autosomal recessive
- Clinical
- Onset: Neonatal to 7 months
- Hypotonia: Generalized; Delayed motor milestones
- Weakness: Proximal > Distal
- Cerebellar: Ataxia; Nystagmus; Dysarthria
- Mental retardation
- Lab
- CK: 900 to 5,000
- Muscle
- Fiber size variation
- Connective tissue: Increased
- Normal dystrophin & merosin
- Cerebellum: Atrophy; Dandy-Walker variant
- White matter: Normal
- Non-progressive
Muscle-Eye-Brain Disorders
19
|
Fukuyama Muscle-eye-brain disease (Santavuori) Walker-Warburg |
- General CNS changes: "Cobblestone" cortex
- Gyral absence or malformations
- Disorganized cortex of variable thickness
- Deficient neuronal migration
- Walker-Warburg Syndromes (WWS): General
- Clinical: Systems with anomalies
- Brain
- Muscle
- Eye
- Gene disorders: Generally produce abnormal glycosylation of α-dystroglycan
- MDDGA1: POMT1; 9q34
- MDDGA2: POMT2; 14q24
- MDDGA3: POMGnT1; 1p34
- MDDGA4: Fukutin: 9q31
- MDDGA5: FKRP; 19q13
- MDDGA6: LARGE1; 22q12
- MDDGA7: ISPD; 7p21
- MDDGA8: GTDC2; 3p22
- MDDGA9: DAG1; 3p21
- MDDGA10: TMEM5; 12q14.2
- MDDGA11: B3GALNT2; 1q42
- MDDGA12: SGK196; 8p11
- MDDGA13: B3GNT1; 11q13
- MDDGA14: GMPPB; 3p21
- MDDGA, Other: Many not identified
- Also see: MDDGC15; DPM3
- Note: MDDGB & MDDGC are
- Alleic with MDDGA (Same genes)
- Milder congenital muscular dystrophies (MDDGB)
- Limb girdle muscular dystrophy syndromes (MDDGC)
- MDDG: Biochemical types
- Dystroglycanopathies
- Other lissencephalies
- Miller-Dieker
- X-linked lissencephaly with agenesis of corpus callosum
- Norman-Roberts
- Miller-Dieker
- Muscle histology
- Myopathy with increased connective tissue
- α-dystroglycan: Hypoglycosylation
- Clinical: Systems with anomalies
- Walker-Warburg Syndrome (MDDGA1)
● O-Mannosyltransferase 1 (POMT1); Chromosome 9q34.13; Recessive
- Genetics: POMT1
- Mutations
- Misense: Gly76Arg
- Stop & Frameshift: Gln303Stop; Gln385Stop; 2110InsG; 2167InsG
- Transversion: 1283T to A,
- Mutations
- Genetic heterogeneity
- POMT1 mutations identified in 20% of Walker-Warburg families: Typical WW syndromes
- Allelic with
- Genetics: POMT1
- O-Mannosyltransferase 1 (POMT1) protein
- O-mannosyl-transferase
- Different isoforms due to alternative splicing
- Ubiquitously expressed
- Peak levels: Adult testis; Skeletal & cardiac muscle; Fetal brain
- Functions
- Synthesizes O-mannose linked core of oligosaccharide with POMgnT1
- POMT1 catalyses first step in O-mannosyl glycan synthesis
- O-Mannosylated proteins
- O-Mannosylated proteins in mammals: α-dystroglycan, N-CAM, tenascin-J1
- O-glycans attached to α-dystroglycan play a role in molecular recognition of laminin-α2
- O-Mannosyl glycan sequences share
- Galβ1-4GlcNAcβ1-2Man-OSer/Thr
- Sialated variant in both brain & muscle: Neu5Acα2-3Galβ1-4GlcNAcβ1-2Man-O-Ser/Thr
- O-Mannose�linked glycosylation locations: Brain, Peripheral-nerve, Muscle glycoproteins
- 30% of all O-linked sugar chains in brain are O-mannose based
- Other disorders of glycosylation: POMGnT1, Fukuyama CMD & FKRP, LARGE1
- O-Mannosylated proteins in mammals: α-dystroglycan, N-CAM, tenascin-J1
- Epidemiology
- World-wide distribution of WW syndrome
- POMT1 mutations identified in Turkish & European families
- Clinical
- Most severe CMD with glycosylation disorder
- Motor: Hypotonia
- CNS
- Seizures
- Severe retardation
- Eye: Multiple anterior & posterior anomalies; Congenital
- Anterior
- Cataract
- Shallow anterior chamber
- Microcornea & microphthalmia
- Lens defects
- Posterior
- Retina: Detachment, Dysplasia & Undifferentiated neurons
- Optic nerve & macula: Hypoplasia & atrophy
- Coloboma
- Anterior
- Other: Testicular defects in males
- Course: Death in fetus or infancy
- Usually < 1 year
- Range 0.1 to 3 years
- Laboratory
- CK: Variable; POMT1 mutation patients > 1,500
- Muscle pathology
- Mild dystrophic changes
- α-Dystroglycan in connective tissue: Reduced glycosylation
- Normal merosin
- CNS pathology
- Cortex
- Migrational defects
- Pathology: Neuronal heterotopia
- Pathophysiology
- Defects in the pial glia limitans
- Result in neural over-migration during neocortex lamination
- Thin
- Hydrocephalus: Dilated ventricles
- Lissencephaly: Type 2; Not classic 4 layered cortex; Cobblestone
- Occipital encephalocoele
- Migrational defects
- White matter: Disorganized myelination; Absent or thin corpus callosum
- Cerebellum: Afolia; Vermal or general hypoplasia
- Brainstem: Small/Absent pyramids
- Spinal cord: Small/Absent Lateral columns; Aberrant anterior root entry zones
- Cortex
- Variant syndrome: Congenital muscular dystrophy with Muscle hypertrophy, Microcephaly & Mental retardation
15
- Epidemiology: Italian children
- Genetics: Recessive inheritance
- Onset
- Birth
- Hypotonia
- Clinical
- Hypotonia
- Joint contractures: Hips; Knees; Ankles: Elbows; Neck flexors
- CNS: Psychomotor retardation; Absent speech
- Motor: Inability to walk; Diffuse weakness
- Muscle hypertrophy: Calf; Quadriceps; Tongue
- Ocular: Normal
- Course: Non-progressive
- Laboratory
- Serum CK: Very high
- Muscle biopsy
- Dystrophic changes
- Small muscle fibers
- Connective tissue increased
- Little degeneration or regeneration
- Laminins
- α2: Partial reduction
- α5: Increased
- Dystrophic changes
- Brain MRI
- Posterior fossa: Enlarged cisterna magna; Cerebellar vermis ± hemisphere hypoplasia
- White matter: Patchy signal, especially periventricular
- EKG: Normal
- Also see: CMD with cerebellar cysts
● O-Mannosyltransferase 2 (POMT2)
- Nosology: Muscular dystrophy-Dystroglycanopathy (Congenital with brain and eye anomalies), Type A, 2
- Epidemiology: > 6 families
- Genetics
- Mutations: Null common; c.1005+1G>A (splice site), Gly185Arg, Arg413Pro, T433X, R638X
- Allelic with
- Congenital muscular dystrophy-dystroglycanopathy with mental retardation B2 (MDDGB2)
- Limb-girdle muscular dystrophy-dystroglycanopathy C2 (MDDGC2; LGMD 2N, R14)
- Congenital muscular dystrophy-dystroglycanopathy with mental retardation B2 (MDDGB2)
- POMT2 protein
- O-mannosyltransferase 2
- Forms complex with protein O-mannosyl-transferase 1 (POMT1)
- Catalyses 1st step in synthesis of O-mannosyl glycan (n α-dystroglycan)
- Short transcript: Widely expressed
- Long transcript: Expressed only in testes
- Subcellular: Associated with endoplasmic reticulum
- O-mannosyltransferase 2
- Clinical: Similar to other WWS syndromes
- Onset
- Age: Prenatal
- Hydrocephalus
- Hypotonia
- Ophthalmic: Cataracts; Microphthalmia
- Onset
- Laboratory
- Serum CK: High
- MRI: Lissencephaly; Hydrocephalus; Hypoplasia of pons & cerebellum
- Muscle biopsy
- Fiber size: Varied
- Endomysial connective tissue: Increased
- Basophilic muscle fibers
- α-dystroglycan staining: Reduced
- POMT2 Variant syndrome: LGMD R14
79
- Nosology: LGMD 2N
- Epidemiology: 30 patients
- Genetics: POMT2 mutations
- Inheritance: Recessive
- Homozygous or Heterozygous
- Missense: All patients with at least one
- Clinical
- Onset
- Age: Birth to 55 years; Median 4 years
- Walking: Late
- Serum: High CK
- Motor
- Weakness
- Proximal > Distal
- Legs > Arms
- Legs: Hip extensors & Knee flexors
- Symmetric: In majority; More asymmetry in adults
- Functional: Slow running
- Calf hypertrophy
- Scapular winging
- Course: Slow progression
- Weakness
- Skeletal: Lordosis, mild
- Intellect: Cognitive impairment in most
- Eye: Normal
- Cardiac: Usually normal; Some with reduced LVEF, bundle branch block
- Onset
- Laboratory
- Serum CK: High; 300 to 5000
- EMG: Myopathic
- Muscle biopsy: Myopathic
- Fiber sizes: Varied
- Necrosis & Regeneration
- Cells: Macrophages
- α-dystroglycan: Reduced, or absent, glycosylation
- Muscle MRI: Involvement of posterior thigh, obturator, paraspinal & glutei
- Brain MRI
- Abnormal (30%): More severely involved patients
- Ventricular enlargement, Periventricular changes
● O-linked mannose β1,2-N-acetylglucosaminyltransferase (POMGnT1)
- Epidemiology
- Disease focus: Finnish (Founder effect)
- Other: Turkish, French, Japanese, Chinese & Korean families
- POMGnT1 gene
- Mutations
- Types: Missense, Nonsense & Frameshift
- More severe phenotype with mutations toward 5� end of gene
- Allelic disorders
- MDDGA3
- Congenital muscular dystrophy, milder (MDDGB3)
- Limb girdle muscular dystrophy (MDDGC3; LGMD 2O)
- Retinitis pigmentosa 76
- Mutations
- POMGnT1 protein function
- O-Mannosyl glycosylation
- Transfers N-acetylglucosamine residue to an O-linked mannose
- O-Mannosyl glycan required for binding between α-dystroglycan & laminin
- Glycosylation stabilizes main O-glycosylation branch
- ? Role in neuronal migration
- Mutation effects: Reduced enzyme function
- O-Mannosyl glycosylation
- Clinical
- Severity: Milder than Walker-Warburg
- Motor
- Hypotonia: Congenital
- Development: Slow
- CNS: Severe mental retardation
- Eye
- Coarse trabecular meshwork in anterior chamber: Myopia (>-6 diopters); Glaucoma
- Retinal dysplasia
- Juvenile cataracts
- Death
- Age: 6 to 42 years
- Longer survival than Walker-Warburg
- Laboratory
- Serum CK: Mild elevation
- Visual evoked potentials: Giant
- Muscle pathology
- Infancy: Muscle fiber degeneration & regeneration
- Dystrophic: Childhood
- Muscle fiber size: Varied
- Muscle fiber regeneration
- Endomysial connective tissue: Increased
- Connective tissue components
- α-Dystroglycan: Reduced
- Merosin: Reduced (to 10% to 20%)
- Laminin-β2: Increased
- CNS pathology
- Brainstem: Thin
- Cerebellar: Hypoplasia with absent inferior vermis; Cysts
- Cortex: Disorganization (cobblestone); Midline defects; Hydrocephalus
- White matter: Abnormal, especially diffuse at young ages < 1.5 years
- POMGNT1 variant syndrome: Muscular dystrophy (MDDGC3; LGMD R15)
- Nosology: LGMD 2O
- Epidemiology: Single Irish patient
- Genetics
- POMGnT1 mutation: Homozygous; Asp556Asn
- Clinical
- Onset age: 12 years
- Weakness
- Proximal > Distal
- Neck
- Hip girdle,
- Shoulder abductors
- Muscle size
- Hypertrophy: Calves; Quadriceps
- Wasting: Hamstrings; Deltoid
- Contractures: Posterior ankles
- Intellect: Normal
- Cardiac: Normal
- Progression: Loss of ambulation < 20 years
- Laboratory
- Serum CK: Very high (> 5,000)
- EMG: Myopathic
- Muscle: Dystrophic; α-dystroglycan staining present with varied intensity
● Isoprenoid synthase domain containing (ISPD)
- Epidemiology: > 10 families; 2nd most common form of WWS (10%)
- Genetics
- Mutations
- Types: Deletions; Missense (ISPD domain); Nonsense
- Effects: Loss of function
- Allelic with
- Congenital Muscular Dystrophy (MDDGB7)
- Limb-Girdle Muscular Dystrophy (MDDGC7)
- Mutations
- ISPD protein
- Mutations impair protein O-mannosylation: 1st step in synthesis of laminin binding glycan
- Clinical syndromes
- Walker-Warburg
- Severe phenotype
- Hypotonia
- Eye: Microphthalmia; Cataracts; Glaucoma; Retinal pathology; Optic nerve hypoplasia
- Death: 80% Neonatal to 24 months
- Muscle-Eye-Brain
- Walker-Warburg
- Laboratory
- Serum CK: Very high; 1,900 to 100,000
- MRI
- Hydrocephalus
- Cobblestone lissencephaly
- Cerebellar hypoplasia
- Kinked brainstem
- Corpus callosum: Hypoplasia
- Muscle
- Dystrophic
- α-Dystroglycan reduced or absent
- Variant ISPD syndrome: Limb-Girdle Muscular Dystrophy (MDDGC7; LGMD R20)
99
- Nosology: LGMD 2U
- Genetics
- ISPD Mutations: Inframe deletion (Val372del); Missense & Stop
- Clinical
- Onset
- Age: Early childhood
- Hypotonia
- Gait disorder
- Gowers' sign
- Weakness
- Proximal > Distal
- Course
- Progressive
- Loss of ambulation: Often ≤ 12 years
- Muscle size: Hypertrophy
- Brain: Normal
- Cardiac: Mildly reduced LV function
- Onset
- Muscle
- Fiber size: Varied
- Endomysial connective tissue: Increased
- Muscle fiber necrosis & Regeneration
- α-dystroglycan: Absent or Severely reduced
- Laminin-α2: Reduced
- Laboratory
- Serum CK: 700 to 9000
- Variant ISPD syndrome: Limb-girdle dystrophy with Cerebellar involvement
- Onset age: < 1 year
- Weakness: Proximal
- Eye
- Myopia: Severe
- Oculomotor apraxia
- CNS: Cerebellar cysts & hypoplasia
- Variant ISPD syndrome: Congential muscular dystrophy (MDDGB7)
70
- Genetics
- Mutations: Missense (Ile153Thr) & Splice (c.535-3C>G, c.1251G>A)
- Clinical
- Weakness
- Hypotonia
- Proximal & Distal
- Non-ambulant
- Respiratory & Face: Normal
- Course: Progressive over 1st year
- Contractures: No
- Cardiac, CNS & Eye: Normal
- Weakness
- Laboratory
- Serum CK: 2000 to 6000
- Muscle
- Fiber size: Varied
- Endomysial connective tissue: Increased
- α-dystroglycan: Reduced or Absent
- Brain MRI: Normal
- Genetics
● Protein O-Mannose β-1,4-N-Acetylglucosaminyltransferase 2 (GTDC2; AGO61; c3orf39; POMGNT2)
- Epidemiology: Consanguineous WWS families
- Genetics
- Mutations: Arg158His; Trp197X; Arg445X
- Allelic disorders: LGMD syndrome
- GTDC2 protein
- Contains uncharacterized glycosyltransferase domain
- High in brain, muscle, heart & kidneys
- Pathway: α-Dystroglycan glycosylation
- Disorders: α-Dystroglycan glycosylation
- Clinical: Walker-Warburg Syndrome
- Brain
- Cobblestone lissencephaly
- Hydrocephalus, severe
- Cerebellar vermis: Hypoplasia
- Eyes: Retinal hypoplasia; Microphthalmia; Macrophthalmia
- Muscle: Hypotonia
- Death: Few months of age
- Brain
- POMGNT2 variant syndrome: Limb-Girdle MD syndrome (MDDGC8)
129
- Nosology: LGMD 24
- Epidemiology: 3 Japanese patients
- Genetics
- Inheritance: Recessive
- Mutations: c.494T>C (Met165Thr) & c.785C>T (Pro253Leu), Heterozygous; c.785C>T (Pro253Leu), Homozygous
- Clinical
- Onset age: 11 mo to 13 years
- Weakness: Proximal; 1 patient
- Calf hypertrophy
- Face: Normal
- CNS: Intelligence reduced in 2 patients
- Laboratory
- Brain MRI: Normal
- Muscle MRI: Diffuse atrophy
- Serum CK: 300 to 4,000
- Muscle biopsy
- Muscle fiber necrosis & regeneration: Scattered
- Internal nuclei
- Endomysial connective tissue: Increased
- Perimysium: Increased cells
- α-Dystroglycan: Absent
● Ribitol Xylosyltransferase 1 (RXYLT1; TMEM5)
- Epidemiology: 13 patients
- Genetics
- Mutations: Nonsense; Frame shift
- Allelic disorder: Congenital MD + CNS
- RXYLT1 protein
- UDP-D-Xylose:Ribitol-5-phosphate β1-4 Xylosyltransferase
- Linkage of 1st Xylose residue to ribitol-5-phosphate
- Exostosin family domain
- Membrane protein (Type 2)
- Role: α-Dystroglycan glycosylation
- RXYLT1 product: Acceptor substrate for B4GAT1 to add Glucuronate moiety
- Mutation: Absent (-3GlcAβ1-3Xylα1-) repeat
- Clinical
- CNS syndromes
- Walker-Warburg
- Muscle-Eye-Brain (MEB)
- Hypotonia
- Retinal dysplasia
- Gonadal dysgenesis
- Course: Death in utero to 2 years
- CNS syndromes
- CNS pathology
- Lissencephaly: Cobblestone
- Occipital neural tube defects
- Pachygyria
- White matter pathology
- Cerebellum: Dysplasia or Atrophy
- Pons: Atrophy
- Laboratory
- Serum CK: Very High
- Muscle
- Myopathy
- Muscle fiber necrosis
- Endomysial connective tissue: Increased
- α-Dystroglycan: Reduced
- TMEM5 variant syndrome: Congenital Muscular Dystrophy + CNS
132
- Epidemiology: 1 Italian male
- Genetics
- Mutation: Homozygous c.139delG
- Clinical
- Onset
- Age: Birth
- Respiratory distress
- CNS
- Developmental milestones: Delayed
- Intellectual disability: Moderate
- Weakness: Mild limb-girdle muscle involvement
- Fatigue
- Cochlear dysplasia
- Course: Stable
- Onset
- Laboratory
- CNS MRI
- Frontotemporal polymicrogyria
- White matter: Abnormal signal
- Cerebellar: Abnormal folia
- Pons: Hypoplasia
- EEG: slow waves
- Serum CK: High; 2,000 to 3,200
- Muscle pathology
- Muscle fiber size: Variation
- Necrosis
- CNS MRI
● Protein O-Mannose Kinase (SGK196; POMK)
- Epidemiology: 6 families
- Genetics
- Mutations: Heterozygous; Missense; Leu137Arg, V302D, Gln258Arg
- Allelic with: MDDGC12
- POMK (SGK196) protein
- Ser/Thr protein kinase family: Kinase probably inactive
- Membrane protein
- ? ATP binding
- α-Dystroglycan glycosylation
- Clinical
- Eye: Hyperplastic primary vitreous; Myopia; Microphthalmia.
- Hypotonia
- Seizures
- IQ: Very reduced
- Meningoencephalocele
- Death: 3 to 4 years
- Laboratory
- Head imaging
- Hydrocephalus: Triventricular
- Cortex: Thin; Agyria; Cobblestone lissencephaly
- Cerebellar hemispheres & Brainstem: Compressed
- Arnold Chiari malformation
- Serum CK: 3,000 to 7,000
- Muscle
- Myopathy
- Normal: Dystrophin; Sarcoglycans; Laminin-α2
- Head imaging
- SGK196 (POMK) variant syndrome: MDDGC12
- Epidemiology: 2 Jordanian siblings
- Genetics
- Mutation: Truncating; Q109X
- Allelic disorder
- Clinical: MDDGC
- Onset age: Infancy
- Weakness
- Infancy: Hypotonia; Delayed walking
- Young adult: Proximal > Distal; Mild face; Independent ambulation
- Calf: Pseudohypertrophy
- Tendon reflexes: Reduced
- CNS: IQ 80 to 85
- Retina: Normal
- Laboratory
- Serum CK: High; 1000 to 1400
- Muscle: Chronic myopathy
- Necrosis & Regeneration
- Dystrophin, dysferlin & sarcoglycans normal
- Brain imaging: Normal or Unrelated focal changes
● β-1,3-N-acetylgalactosaminyltransferase 2 (B3GALNT2)
- Epidemiology: US, Europe, Turkey, Saudi Arabia; 6 patients
- Genetics
- Mutations: Heterozygous or homozygous; Missense or Stop
- B3GALNT2 protein
- Location: Endoplasmic reticulum
- Function
- Walker-Warburg phenotype
- Clinical
- Onset age: Birth
- Hypotonia: No motor milestones
- Cognitive development: Absent
- Epilepsy
- Eye: Optic nerve hypoplasia; Cataracts; Microphthalmia
- Laboratory
- MRI: Hydrocephalus; Lissencephaly, Cobblestone
- Serum CK: 1,000 to 21,000
- Clinical
- Muscle-Eye-Brain disease (Fukuyama congenital muscular dystrophy) phenotype
- Clinical
- Onset age: Birth to 17 months
- Delay: Motor & Cognitive
- Eye: Optic nerve hypoplasia or No changes
- Laboratory
- MRI: Polymicrogyria; Leukoencephalopathy; Cerebellar cysts or dysplasia
- Serum CK: 800 to 2,000
- Clinical
- Varied fiber size
- Regeneration
- Endomysial connective tissue: Increased
- Internal nuclei
- α-Dystroglycan: Glycosylation reduced (IIH6 binding variably reduced)
● β-1,3-N-acetylglucosaminyltransferase 1 (B3GNT1; B4GAT1)
- Epidemiology: East Indian & Saudi families
- Genetics
- Mutations
- Homozygous
- Both Asn390Asp & Ala406Val; c.821_822insTT (p.Glu274Aspfs*94)
- Mutations
- B3GNT1 protein
- Acceptor from RXYLT1: Adds Glucuronate moiety to α-dystroglycan
- Xylose β1,4 glucuronyltransferase
- Forms GlcAα1-4 linkage
- Complex with LARGE1
- Clinical
- Onset age: Prenatal
- Hydrocephalus: Intrauterine
- Walker-Warburg syndrome
- Motor & Cognitive impairment
- Seizures
- Spasticity
- Optic nerve: Agenesis
- GU: Undescended testes, Micropenis, Hydronephrosis
- Death < 1 year
- Laboratory
- Serum CK: High, 3,180 to 15,000
- MRI
- Ventricular enlargement
- Diffuse widening of gyri
- Disorganization of cortical sulci: cobblestone lissencephaly along posterior occipital & temporal lobes
- Muscle
- Reduced laminin-α2 & α-sarcoglycan
- α-dystroglycan: Reduced glycosylation; Reduced binding of to laminin
- Brain pathology
- Lissencephaly type II: Cortical dysplasia, Glio-neuroepithelial leptomeningeal heterotopia
- Obliteration of subarachnoid space
- Communicating hydrocephalus
- Cerebellum: Cortical dysplasia/hypoplasia; Inferior vermian defect
- Pyramids: Medulla hypoplasia
- Inferior olives: C-shaped dysplasia
- Hydromyelia
- Retina: Focal disorganization; Dysplasia
● GDP-Mannose Pyrophosphorylase B (GMPPB)
- Nosology: LGMD 2T
- Epidemiology: 8 families
- Genetics
- Mutations: Homozygous or Heterozygous; Missense (Most common) or Stop (Severe patient)
- GMPPB protein
- Mannose-1-phosphate guanyltransferase beta
- Location: Cytoplasm
- Catalyzes: α-D-mannose-1-phosphate + GTP → GDP-α-D-mannose
- GDP-mannose: Required in 4 glycosylation pathways
- Inhibited by: GMPPA
- Clinical
- Allelic syndromes
- Walker-Warburg (MDDGA14)
- Congenital MD (MDDGB14)
- LGMD 2T (MDDGC14)
- Myasthenic syndrome
- Walker-Warburg (MDDGA14)
- Onset age: Birth to 4 years
- Weakness
- Hypotonia
- Poor head control
- Delayed walking (80%)
- Difficulty climbing stairs
- Feeding difficulties (25%)
- Exercise intolerance: Milder patient
- CNS
- Intellectual delay (80%)
- Epilepsy (40%)
- Eye: Cataracts (40%); Retinal disease (10%)
- Cardiac: Cardiomyopathy (25%)
- Allelic syndromes
- Laboratory
- Serum CK: High; Range 630 to 7300
- Brain MRI: Cerebellar hypoplasia (40%)
- Muscle: α-dystroglycan has reduced glycosylation
- GMPPB variant syndrome: Limb Girdle Muscular Dystrophy 2T (MDDGC14; LGMD 2T)
- GMPPB genetics
- Inheritance: Recessive
- Mutations: Missense; Asp27His, Arg287Gln, Pro32Leu, Cys266Tyr
- Clinical
- Onset age: Birth to 4th decade
- Weakness
- Limbs, proximal
- Course: Slow progression
- Rhabdomyolysis & Cramps
- Muscle size: Enlarged calves in some patients
- CNS: Learning difficulties (50%)
- Cardiomyopathy: 1 patient
- Laboratory
- Serum CK: High
- Muscle
- Fiber size: Varied
- Internal nuclei
- Muscle fiber necrosis
- Endomysial connective tissue: Increased
- α-dystroglycan: Reduced
- Western blot: Laminin-α2 reduced
- Muscle MRI: Involvement of Paraspinals, Hamstrings & Medial Gastrocnemius
- GMPPB genetics
- GMPPB variant syndrome: Myasthenic syndrome
122
- Epidemiology: 7 patients in 5 families
- Genetics
- Inheritance: Recessive
- Mutations: All through gene; Missense in at least 1 allele
- Clinical
- Onset age: 1.5 to 31 years
- Weakness
- Proximal limbs
- External rotation: Arm & Thigh
- Variable: Activity-dependent fatigue; Improve with rest
- Face & Eyes: Spared
- Treatment: Pyridostigmine ± Salbutamol
- Cognition: Normal, or mildly delayed
- Laboratory
- EMG: Myopathic
- RNS: Decrement in proximal muscles
- SFEMG: Abnormal
- Serum CK: High; 400 to 3000
- Muscle pathology: Myopathic; α-Dystroglycan reduced
- Muscle MRI: Proximal > Distal involvement
Congenital Muscular Dystrophy with Familial Junctional Epidermolysis Bullosa (EBS5B)
157
●
Plectin (Plec)
- Genetics
- Gene structure
- 32 exons
- Exons 2 to 32: Constant
- First exons
- 8 alternative
- Variable length & sequence
- Spliced into a common exon 2
- Mutations
- Types: Missense, Stop, Amall deletions & duplications
- Location: Exon 31 (Rod domain) common
- Allelic disorders
- CMD + Epidermolysis Bullosa (EBS5B)
- LGMD with no Skin changes (LGMD 2Q; LGMD17)
- EBS-MD + Myasthenic syndrome (EBS-MD-MyS) ± Scapuloperoneal myopathy, Late-onset
- EBS + Pyloric atresia (EBS5C)
- EBS5A, Ogna type
- EBS, generalized (EBS5D)
- Hearing loss, non-syndromic or auditory neuropathy spectrum
- Gene structure
- Plectin protein
- Size: 500-kD
- Structure: Contains intermediate filament & actin-binding domains
- Family: Plakin
- Tissue expression: Ubiquitous; Highest in stratified squamous epithelia, muscle & brain
- Subcellular location: Concentrated at sites of stress
- NMJ: Postsynaptic membrane
- Muscle: Sarcolemma & Z-disks
- Skin: Hemidesmosomes
- Heart: Intercalated disks
- Complexed with
- Desmuslin
:
Associated with α-dystrobrevin
- Desmin
- Other: Microtubules, α-Spectrin, Nuclear lamin B, Integrin β4, Microtubule-associated proteins
- Desmuslin
- Functions
- Plectin types
- 1: Connects Desmin intermediate filaments to Nucleus/ER
- 1B
- Links Desmin intermediate filaments to mitochondria
- Reduction: Mitochondrial shape changes
- 1D
- Binds plectin to sarcomere
- Muscle & Heart specific
- 1F
- Tethers Desmin intermediate filaments to sarcolemma
- Location: Sarcolemma
- Other associations: Dystrophin; β-Dystroglycan
- Cytoskeletal linker: Connects
- Intermediate filaments
- Actin
- Microtubules
- Organelles
- β4-Integrin
- Type XVII collagen (BP180)
- Nesprin-3a
- Sarcolemmal proteins
- Mechanical integrity of cells: Formation of intermyofibrillar desmin cytoskeleton
- Substrate for caspase 8 during CD95- & tumor necrosis factor receptor-mediated apoptosis
- Plectin types
- Disease association: Partial, but reduced, expression associated with milder disease
- Clinical
- Onset
- Age: Congenital
- Weakness or Hypotonia
- Skin lesions: Blistering
- Skin
- Blistering: Due to Trauma & Heat
- Alopecia
- Muscle
- Weakness: Myopathy & Myasthenic syndrome
- Distribution
- Proximal
- Distal
- Facial
- Ocular
- Course
- Onset: Congenital or Childhood
- Slow progression
- Many in wheelchair by 10 years
- Degree of weakness: May relate to level of plectin expression
- Distribution
- Diffuse muscle hypotrophy
- Weakness: Myopathy & Myasthenic syndrome
- Systemic
- Elbow contractures
- Infantile respiratory complications
- Alopecia
- Decayed teeth
- Laryngeal webs
- Urethral strictures
- Cardiomyopathy, Dilated, Biventricular
- CNS: Brain atrophy; Enlarged ventricles
- Treatment
- 3,4-diaminopyridine: Some improvement
- Pyridostigmine: No benefit
- Onset
- Laboratory
- Serum CK: High; > 1,000
- Electrophysiology
- Repetitive Nerve Stimulation: Decrement
- EMG: Myopathic; Irritable
- Endplate physiology
- Quantal release by impulse: Normal
- MEPPs: Small
- AChRs: Fetal & Adult AChRs at NMJs
- Muscle pathology
- Myopathic
- Necrosis & Regeneration
- Muscle fiber size: Varied
- Internal nuclei
- Connective tissue: Increased
- Abnormal internal architecture on NADH stain
- Spike-like sarcolemmal projections with Integrin β1D stain
- Endplates: Chains of small regions over fiber surface
- Plectin
- Absent or reduced staining in muscle Z-lines & skin
- Abnormal related proteins: BP180; BP230
- Hemidesmosomal protein HD1: Reduced staining
- Desmin: Focal deposits in most muscle fibers
- Some patients: Rods; Cytoplasmic bodies; Rimmed vacuoles
- Mitochondria
32
- Increased number of COX- muscle fibers
- Biochemistry: Reduced COX I (Complex I) & cytochrome c oxidase (Complex IV) activities
- Morphology: Abnormal shape; Inclusions; Proliferation
- Also see: Congenital muscular dystrophy with mitochondrial structural abnormalities
- Myopathic
- Plectin Variant syndrome: Limb-Girdle Dystrophy R17 (LGMD R17)
84
- Nosology: LGMD 2Q
- Epidemiology: Turkish & Russian families; 13 patients
- Genetics
- Inheritance: Recessive
- Mutations
- Homozygous
- Location: Exon 1f (Isoform-specific sequence of plectin isoform 1f)
- c.1_9del (Contains initiation codon); Glu20X
- Other mutations with LGD phenotype: Q1022X; G3835S
- Plectin protein: 1f isoform
- Sarcolemma
- More on type 2A muscle fibers
- Costameres
- Interacts with: Dystrophin; β-Dystroglycan
- Clinical
- Onset
- Age: Early childhood
- Delayed walking
- Weakness
- Generalized: Proximal > Distal
- Face & Eyes: Ptosis in few patients
- Course
- Stable in childhood
- Progressive in teens
- Loss of ambulation: Adults
- Muscle sizes
- Atrophy: Axial
- Hypertrophy: Gastrocnemius, Deltoid, Triceps
- Skin: Normal
- Joints: Contractures late
- Pulmonary: Dyspnea; Bronchiolitis & Atelectasis
- Course: Progression in adults
- Onset
- Laboratory
- Serum CK: 3000 to 5500
- EMG: Myopathic
- RNS: Normal or Decrement
- Muscle MRI
- Chest CT: Reticulo-nodular pattern
- Muscle pathology
- Dystrophic
- Fiber size: Varied
- Myonuclei: Internal; Clustered
- Necrosis & Regeneration
- Endomysial connective tissue: Increased
- Type 1 or 2 predominance
- Desmin aggregates
- Myf5-positive nuclei: Increased
- COX- fibers: Some patients
- Plectin
- Reduced on sarcolemma (30%)
- Isoform 1f: Reduced to 1%
- Aggregates
- Ultrastructure
- Membrane duplications
- Enlarged space between membrane & sarcomere
- Misalignment of Z-disks
- ? Disrupted connection between the sarcolemma and sarcomere
- Dystrophic
Congenital muscular dystrophy with Mitochondrial Structural Abnormalities (Megaconial) (MDCMC)
2
●
Choline kinase beta (CHKB)
- Epidemiology: > 40 families; More common in Turkey & Asia
- Genetics
- Mutation types: Nonsense, Missense, Deletion, Splice site
- Locations: All exons except 10
- Nonsense mutations: More severe disease
- Allelic disorders
- Mouse disorder: Rostrocaudal muscular dystrophy (rmd)
- CHKB protein
- Kennedy pathway 186: Synthesis of phosphatidylcholine & phosphatidylethanolamine
- Catalyzes
- Phosphorylation of choline by ATP
- 1st step in phosphatidylcholine biosynthesis
- Mutations
- Choline kinase activities: Absent
- Phosphatidylcholine levels: Decreased
- DRP1 (Mitochoindrial fission protein): Reduced
- Related proteins & disorders: Kennedy pathways for membrane synthesis
- Clinical
- Onset
- Age: Congenital or Early childhood
- Hypotonia
- Motor
- Weakness: Proximal; Slowly progressive
- Hypotonia: Neonatal
- Walking: Late or Never (20%)
- Muscle
- Wasting
- Rhabdomyolysis: Few patients
- Heart (40%)
- Cardiomyopathy
- Type: Dilated
- Common couse of early mortality
- Congenital defects
- Cardiomyopathy
- CNS (100%)
- Developmental delay: Global
- Intellectual disability: Severe
- Autism
- Seizures (30%)
- Head
- Circumference: Small (45%)
- Face: Dysmorphic (50%)
- Skin: Ichthyosis (30% to 80%)
- GI: Pancreatitis; Abdominal pain
- Course
- Often progressive
- May be static
- Episodic exacerbations with illness: Some patients
- Differential diagnosis
- Onset
- Laboratory
- CK: Mildly elevated; 190 to 2700
- Muscle pathology
- Fiber size variation
- Necrosis & Regeneration
- Endomysial connective tissue: Increased
- Mitochondria
- Mitochondria: SDH & COX stain
- Increased at periphery of type 2 muscle fibers
- Reduced in center of fibers (Normal SR in these regions)
- Enlarged
- EM: Enlarged mitochondria with abnormal structure
- Mitochondria: SDH & COX stain
- Lipid: Normal
- Laminin-α2: Normal
- Brain MRI: Often normal
- CHKB variant: Teen onset weakness
135
- Epidemiology; Bulgarian & British patients
- Genetics
- Mutation: Missense; Homozygous; Ser234Leu
- Clinical
- Onset age: 16 years
- Weakness
- Arms & Legs
- Respiratory
- Proximal
- Symmetric
- Intellectual disability: Mild; Mutism
- Skin: Normal
- Laboratory
- Serum CK: 600 to 1,500
- EMG: Myopathy
- Muscle MRI
- Thigh: Sparing of adductor longus
- Leg: Sparing of extensor digitorum longus
- Muscle
- Mitochondria
- Size: Very large & elongated
- Location: Predominantly at muscle fiber periphery
- Oxidative enzyme complexes: Normal
- Aggregates: p62 & LC3 in some patients
- Mitochondria
Congenital Muscular Dystrophy with early Spine Rigidity (CMYO3)
●
Selenoprotein N, 1 (SEPN1; SELENON)
|
|
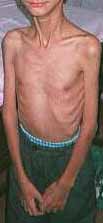
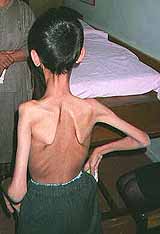
Congenital muscular dystrophy with respiratory failure & muscle hypertrophy (CMD1B; MDC1B)
7
●
Chromosome 1q42; Recessive
- Epidemiology: Arab & German families
- Clinical
- Severe diaphragm involvement: Respiratory failure
- Weakness
- Neck
- Proximal > Distal
- Arms & Legs
- Face: Mild
- Walking: Onset at 1.5 to 2.5 years
- No clear progression of weakness
- Muscle hypertrophy: Generalized
- Contractures
- Ankles
- Spine rigidity (50%)
- Intellect: Normal
- Laboratory
- Serum CK: Very high; 1,700 to 7,600
- Brain: Normal MRI
- Muscle
- Dystrophic changes
- Connective tissue proteins: Reduced on some fibers
- Merosin: Reduced; No mutations in Merosin gene
- Integrins α-7 & β1D: Reduced
- Heparan sulfate proteoglycans: Normal
CMYO10A: Early-onset Myopathy with Areflexia, Respiratory Distress & Dysphagia
87
●
Multiple epidermal growth factor-like domains 10 (MEGF10)
- Nosology: EMARDD; CMYO10A; CMYP10A
- Epidemiology: > 20 patients
- Genetics:
- Mutations: Often truncating; Missense Cys699Ser, Cys774Arg
- Allelic disorder: Congenital myopathy with minicores
- Transmembrane protein
- Multiple epidermal growth factor family
- Possible functions
- Cell-cell adhesion
- Engulfment receptor of apoptotic cells: Normal clearance of apoptotic neurons during neurogenesis
- Regulation of muscle development & repair
- Promotion of muscle precursor proliferati
- Suppression of myoblast differentiation.
- Likely mediated through Notch signaling pathway
- Expression
- CNS: Brain, Spinal cord (Interneurons & motor neurons in spinal gray matter)
- Skeletal muscle & NMJ: Satellite cells
- Onset age: Early or Prenatal with reduced fetal movements
- Hypotonia: Motor milestones delayed
- Weakness: Proximal & Distal
- Skeletal
- Contractures: Fingers, Elbows & Equinovarus foot
- Scoliosis
- Areflexia
- Respiratory
- Distress & Failure: Paradoxical breathing
- Diaphragm eventration or paralysis (Unilateral or Bilateral)
- Dysphagia: Feeding often by gastrostomy
- Cognition: Normal
- Course
- Slow progression
- 50% die < 15 years
- Serum CK: Normal to 700
- EMG: Myopathic; Normal RNS & NCV
- Muscle
- Muscle fiber size: Reduced; Mildly varied
- Connective tissue: Reduced numbers of cells
- MEGF10 protein: Absent in all muscle fibers
- Myofibrillar: Cytoplasmic inclusions; Minicores
- No detectable PAX7+ nuclei
- Sural nerve: Normal
- Nosology: CMYO10B; CMYP10B
- General: Less severe than EMARDD
- Epidemiology: 2 families
- Genetics
- Inheritance: Recesssive
- Missense MEGF10 mutations; C326R, C774R
- Clinical
- Onset
- Age: Infancy
- Weakness: Hypotonia; Neck
- Weakness
- Proximal & Distal
- Neck: Flexion
- Ankles: Dorsiflexion > Plantarflexion
- Difficulty walking & climbing stairs
- Respiratory: Require BiPAP at night; Onset 10 years
- Progression
- To teens; Then static
- Loss of ambulation: Some in teens
- Tendon reflexes: Reduced or Absent
- Scoliosis: Onset 1st decade
- Contractures: Neck; Elbows; Knees; Heels
- GI: Reflux; High arched palate
- Cardiac: Normal
- Onset
- Laboratory
- Serum CK: Normal or Mildly high
- Muscle biopsy
- Myopathic
- Fiber size: Varied: Large & small
- Emdomysial connective tissue: Increased;
- Fatty replacement
- Internal nuclei
- Generally inactive
- Internal architecture: Motheaten or Minicores
- Ultrastructure: Focal Z-band disarray across few sarcomeres
- Myopathic
Congenital Muscular Dystrophy with Muscle hypertrophy (MDC1C; MDDGB5)
16,
26
●
Fukutin-related protein (FKRP)
- Epidemiology: > 50 families
- Genetics
- Allelic with
- LGMD 2I (MDDGC5)
- Myopathy with abnormal merosin (Laminin-2)
- Walker-Warburg syndrome (MDDGA5)
: Homozygous FKRP mutations; Met1Val & Cys318Tyr
- LGMD 2I (MDDGC5)
- Mutations: Missense
- Ser221Arg, Ala455Asp, Pro448Leu, Tyr309Cys, Ser385Ter, Pro316Thr, Tyr465Ser
- Tyr465Ser mutation adds potential new glycosylation site
- No patients with 2 FKRP null alleles
- Patients with L276I mutation usually have LGMD 2I
- Allelic with
- FKRP protein
- Ubiquitous
- Highest levels in skeletal muscle, heart & placenta
- Subcellular: Normal protein in Golgi
- Function: α-Dystroglycan glycosylation
- More severe cases: Mutant protein
54
- Location: Endoplasmic reticulum
- Shorter half-life
- Preferentially degraded by proteasome
- Clinical
- Onset
- First weeks of life
- Hypotonia
- Reduced movement & activity
- Weakness
- Diffuse: Limbs; Neck; Trunk; Face
- Functional abilities
- May sit unassisted
- Inability to walk
- Muscle size
- Hypertrophy: Especially calf; Most patients
- Wasting: Shoulder girdle; Other muscles
- Contractures
- Some patients
- Knees; Ankles; Elbows; Spine
- Skeletal: Small head; Scoliosis
- CNS
- Seizures
- Some patients with
- Mild mental retardation: Psychomotor; Absent speech
- Cerebellar cysts
- Onset
- Laboratory
- Serum CK
- Very high: 3,000 to 8,000
- May have lower levels with increasing age
- EMG: Myopathic
- Muscle biopsy
- Varied fiber size
- Connective tissue: Increased
- Necrosis & Regeneration: Occasional fibers
- Type I predominance: Mild
- Laminins
- α2: Partial reduction around muscle fibers; Normal in nerve
- α5: Increase
- Dystroglycans
- α-dystroglycan
- Muscle staining: Marked reduction: Similar to changes seen in Fukuyama CMD
- Western blot: Abnormal molecular weight
- β-dystroglycan: Normal
- α-dystroglycan
- Brain MRI: Normal or Cerebellar cysts
- EKG: Normal
- Serum CK
- Also see: CMD with muscle hypertrophy, mental retardation & cerebellar hypoplasia
Ullrich Congenital Muscular Dystrophies 1 (UCMD1; Scleroatonic) 21
● UCMD1A● UCMD1B
● UCMD1C
- Genetics
- Mutations
- Recessive (COL6A1, COL6A2 & COL6A3)
- Truncating or Missense
- Reduced or absent Collagen VI in muscle
- Commonly: Located in triple helical domain; Alter glycine residues
- Dominant (COL6A1, COL6A2 & COL6A3)
- Inframe deletion or mutation
- Dominant negative effect 41: Interferes with dimer formation & secretion of molecule
- Often in N-terminal region: Proximal to cysteine in triple helical domain
- Family history of disease uncommon
- Recessive (COL6A1, COL6A2 & COL6A3)
- Allelic disorders
- Some patients with similar syndrome
- Not linked to these loci: Normal Collagen VI
- See: COL12A1
- Mutations
- Collagen VI protein
- Ubiquitious extracellular matrix protein
- Concentrated outside basal lamina
- Ullrich myopathy muscle & fibroblasts
- Mislocalized, Low or Absent
- Clinical features
- Variable
- Some with severe weakness
- Some families with COL6A3 mutations have milder disease
- Onset
- May be reduced fetal movements
- Congenital contractures
- Hypotonia
- Joints
- Early
- Hyperlaxity of distal joints
- Distal arthrogryposis: Some patients
- Contractures
- Proximal > Distal
- Knee contractures may limit walking
- Spine: Rigidity; Kyphoscoliosis
- Torticollis
- May improve with increasing age
- Weakness
- Diffuse: Distal > Proximal; Neck flexors
- Ambulation
- May walk by age 1 to 2 years (30%)
- Majority never walk
- Respiratory insufficiency
- Onset: Hypoventilation in 1st decade
- General
- Mild disease: VC &g; 60% of predicted
- More severe disease: VC &l; 50% of predicted
- Failure: Not strongly correlated with degree of weakness
- Muscle wasting: Distal predominant
- Course
- Slowly progressive
- Walking
- Some walk for a limited time: 3 to 6 years
- Death
- Age: 1st or 2nd decade
- Associated with respiratory failure
- Skin
- Soft
- Keratosis pilaris
- Keloids
- Atrophic scars
- Striae
- Petechiae
- Skeletal
- Prominent calcaneus of foot
- Scoliosis
- Rigid spine
- Tendons: Similar to Ehlers-Danlos; Contractures
- Cardiac: Normal
- Intelligence: Normal
From: A Connolly MD
Hyperkeratosis pilari - Early
- Variable
- Laboratory features
- Serum CK: Normal to 10x elevated
- EMG: Myopathic
- Muscle biopsy
- Varied muscle fiber size: Some very small muscle fibers
- Endomysial connective tissue: Progressive increase
- Necrotic muscle fibers: Rare or Occasional
- Lobulated fibers: Occasional
- Collagen expression
- Type VI
- 2 patterns of pathology
- Absent from skeletal muscle & capillaries
- Absent on surface of muscle fibers but present in connective tissue
- No correlation between pattern of pathology and clinical phenotype
- 2 patterns of pathology
- Type IV: Upregulated
- Type VI
- Ultrastructure
- Muscle plasma membrane: Uneven, extension & folding
- Basal lamina: Thickening & overproduction; Replication of capillary basement membrane
- Capillary morphology 42: Small lumens; Increased pinocytotic vesicles in endothelial cells
- Collagen: Disorganized fibrils
- Gene testing: Utah
Congenital Muscular Dystrophy with Mental Retardation & Abnormal Glycosylation (MDDGA6; MDC1D)
43
●
LARGE1 (Acetylglucosaminyltransferase-like protein)
- Epidemiology: Few patients
- Genetics
- 5th largest human gene
- Mutations
- Missense: Glu509Lys
- Insertion (1bp): 1999insT
- Deletion: Intron 8 to Intron 10
- Allelic with
- Congenital Muscular Dystrophy (MDDGB6)
- Congenital Muscular Dystrophy (MDDGB6)
- Other mutations in: myd mouse
- LARGE1 protein
- Cell location: Golgi
- Ubiquitously expressed
- Highest levels in heart, brain & skeletal muscle
- Two putative catalytic domains with homology to
- α-glycosyltransferase (β1,3-GlcA) (bacterial WaaIJ family)
- Synthesis of outer membrane lipopolysaccaride
- β-1,3-N-acetylglucosaminyltransferase (iGnT)
- Synthesis of poly-N-acetyllactosamine backbone of erythrocyte i antigen
- α-glycosyltransferase (β1,3-GlcA) (bacterial WaaIJ family)
- Role in glycosylation of α-dystroglycan
47
- Glucuronosyltransferase
- Specific to α-linked Xylose
- Generates -3GlcAβ1-3Xylα1- repeat with additional GlcAβ1-4Xylβ1-4Rbo5P unit
- Units are laminin binding glycan domain
- Clinical
- Onset
- Age: 5 months
- Global developmental delay
- Congenital muscular dystrophy
- Hypotonia
- Slow improvement over 1st decade
- Muscle hypertrophy
- Moderate
- Quadriceps; Calf; Arm muscle
- Tongue: Normal
- Muscle atrophy: None
- Weakness
- Proximal > Distal
- Legs > Arms
- Face: Mild weakness
- EOM: Normal
- CNS
- Mental retardation: Profound
- Mirror movements
- Nystagmus: Lateral
- Spasticity: Tone increased; Tendon reflexes increased; Extensor plantar
- Contractures
- Mild
- Ankle & Elbow
- Onset
- Laboratory
- Serum CK: High; 400 to 4500
- NCV: Normal Motor & Sensory
- EMG: Myopathic
- Electroretinogram: Small evoked response
- CNS imaging
- White matter changes: Periventricular to arcuate fibers; Anterior & Temporal
- Structural abnormalities: Abnormal neuronal migration; Small brainstem
- Pathology: Muscle
- α-dystroglycan
- Immunolabelling: Reduced
- Immunoblotting with to glycosylated epitope: Reduced molecular weight of α-dystroglycan
- Retains some laminin binding activity
- Laminin α2: Normal
- Dysferlin: Upregulated in myd mouse
- α-dystroglycan
- Mouse model: myd mouse muscle
Congenital Muscular Dystrophy + Cataracts & Intellectual Disability (MDCCAID)
137
●
Inositol polyphosphate-5-phosphatase K (INPP5K; SKIP)
- Epidemiology: 12 families, 17 patients
- Genetics
- Mutations
- Missense & Stop
- Ile50Thr, Met93Val, Val23Met, p.Gly140Ser, Asp269Asn, Tyr300Cys, Ile363Thr
- No homozygous stop
- Mutations
- INPP5K protein
- Expression: High in brain & muscle
- Cell location: Endoplasmic reticulum; Actin ruffles in cytoplasm
- Catalyzes removal of 5-phosphate from PtdIns(4,5)P2
- Regulate: Myoblast differentiation; NMJ formation
- Myogenic loop triggered by insulin-growth factor II (IGF-II)
- PI3K-AKTmTOR pathway
- Protein processing: Interaction with ER chaperone HSPA5/BiP
- PtdIns(3,4,5) P3-dependent regulation of insulin signaling & glucose homeostasis in skeletal muscle
- Phosphoinositide (PIP phosphatase) disorders
153
- Hereditary Polyneuropathies
- Congenital Myopathy
- INPP5K: Congenital Muscular Dystrophy with Cataracts
- MTM1: X-linked centronuclear myopathy (XLCNM)
- MTMR14: CNM1 modifier
- Also see: Hemifacial Myohyperplasia
- CNS
- Other
- INPPL1
: Opsismodysplasia
- INPPL1
- pPtdIns/PIP kinase disorders
- Clinical
- Onset age: Early childhood
- Muscle
- Development
- Hypotonia
- Motor milestones: Delayed
- Weakness: Moderate to Severe
- Wheelchair: Most
- Development
- CNS
- Intelletual disability
- Spasticity 60%
- Seizures 60%
- Microcephaly, mild
- Skeletal
- Hyperlordosis
- Short stature
- Contractures: Limbs or Spine
- Eye
- Cataract 60%: Congenital
- Strabismus 60%
- Course: Very slow progression
- Laboratory
- EMG: Myopathic in legs
- Serum CK: High; 3x to 14x upper limit of normal
- Muscle MRI (T1): Atrophy & fatty degeneration in proximal limbs
- Muscle pathology
- Fiber size: Varied
- Fiber types: Variable among fascicles
- Endomysial connective tissue: Increased
- Necrosis & Regeneration: Occasional
- Vacuoles: Some patients
- α-Dystroglycan: Reduced or Normal
- Nuclei: Abnormal structure
- Brain MRI: Normal or Atrophy
- Differential diagnosis: Marinesco-Sjögren syndrome
Congenital Muscular Dystrophy with Adducted Thumbs, Ophthalmoplegia & Mental retardation 34
● Autosomal Recessive- Epidemiology: Consanguinous Sicilian siblings
- Onset: Congenital
- Clinical
- Joints: Adducted thumbs
; Toe contractures; Hyperlordosis
- Motor: Hypotonia; Delayed walking
- Weakness: Diffuse; Facial; Severe
- Ocular: Ophthalmoplegia; Ptosis
- Cognitive: Mental retardation, IQ 65 to 70
- Joints: Adducted thumbs
- Laboratory
- Serum CK: High, 130 to 750
- NCV: Normal
- MRI: Cerebellar hypoplasia, Mild
- Muscle
- Dystrophic: Varied fiber size; Increased endomysial connective tissue
- Normal laminins & α-dystroglycan
CDG: Multisystem Disorder 131
● Phosphatidylinositol glycan anchor biosynthesis Class Y protein (PIGY)- Epidemiology: Australian families
- Genetics
- Mutation: c.137T>C (p.Leu46Pro)
- Allelic disorders
- Development delay & Microcephaly (Promoter variant (c.-540G>A))
- Hyperphosphatasia with mental retardation syndrome 6 (HPMRS6)
- PIGY protein
- Glycosylphosphatidylinositol (GPI) anchor biosynthesis
- Clinical
- Onset age: Congenital
- Necrotizing enterocolitis
- Chronic lung disease
- Dysmorphic
- Skeletal
- Digits: Brachyphalangy; Syndactyly
- Arthrogryposis: Elbow & knee flexion contractures; Hip dysplasia
- CNS
- Hypotonia
- Seizure disorder with multifocal spike & slow wave activity
- Development: Global delay; Regression
- Death: 7 months to 2 years
- Laboratory
- Serum CK: 200 to 4,000
- Muscle
- Fiber size: Varied
- Muscle fibers: Necrosis & regeneration; Internal nuclei
- Endomysial connective tissue: Increased
Congenital muscular dystrophy & myasthenic syndrome 55
● DOK7- Epidemiology: Palestinian family, consanguinous
- Genetics
- DOK7 mutation: c.957delC Homozygous
- Allelic with: Limb-Girdle Myasthenia
- Onset
- Age: Neonatal or in utero
- Reduced fetal movements
- Hypotonia
- Clinical
- Motor
- Early: Hypotonia; Reduced anti-gravity movements
- Weakness
- Proximal > Distal
- Most severe: Trunk & Shoulder girdle muscles
- Mild to moderate involvement: Face, Neck, Proximal limb
- None: Extra-ocular, Bulbar
- Course: Slow progression
- Episodic exacerbations: No precipitating factors
- Muscle size: Small, No hypertrophy
- Course
- Some death in childhood or adult: 50% with respiratory infections
- Motor: Static
- Most survivors able to walk as children & adults
- Joints
- No contractures
- Mild laxity in adults
- Intelligence: Normal
- Motor
- Laboratory
- Serum CK: Normal or Minimally elevated
- Brain CT: Normal
- Echocardiogram: Normal
- Muscle pathology
- Morphology: "Dystrophic"
- Normal staining: Laminins (α2, α5, β1, β2), Dystrophin, Sarcoglycans (α, β, γ), α & β-Dystroglycan
Congenital disorder of Tendons, Bone, Muscle & Eyes 141
● Procollagen-proline, 2-oxoglutarate-4-dioxygenase, alpha subunit, isoform 1 (P4HA1)- Epidemiology: 1 Chinese family
- Genetics
- Mutations: Heterozygous; Splice donor (c.1543+2 T>G) & Stop (c.1323_1324insAG)
- P4HA1 protein
- Collagen prolyl 4-hydroxylases
- Location: Lumen of endoplasmic reticulum
- Collagen synthesis
- Role in formation stabilization of triple helical domain of collagens
- External link
- P4HA1
- Expressed in all tissues
- Exon 9 splice form: Major form expressed in muscle tissue, especially < 4 years
- Collagen prolyl 4-hydroxylases
- Clinical
- Onset age: Congenital
- Skeletal
- Joints
- Hypermobility: Distal
- Contractures: Prenatal; Camptodactyly
- Feet: Flat arches; Prominent calcanei
- Face: Dysmorphic
- Bone dysplasia
- Joints
- Muscle
- Hypotonia: Congenital; Distal > Proximsl
- Motor development: Delayed
- Weakness
- Face
- Eyes: Reduced upgaze; Ptosis
- Trunk & Neck
- Limbs: Proximal & Distal
- Gait: Waddling
- Eye: High myopia
- Cognition: Normal
- Course: Improvement of motor function over time
- Laboratory
- Serum CK: 358
- Muscle MRI: Legs diffusely increased T1 signal; Thighs Vastus lateralis involvement
- EMG: Normal
- Muscle biopsy
- Fiber size: Small; Especially type 1
- Endomysial connective tissue: Increased
- Collagen IV & Laminin-γ1: Reduced in basement membrane
- Bones: Osteopenia, diffuse
Intellectual Disability, Hypotonia, Motor Neuropathy & Deafness (NEDHND; CMND)
●
β-IV-Spectrin, Nonerythrocytic (SPTBN4; Spectin β-IV)
- Epidemiology: > 30 patients
- Genetics
- Mutations: Stop, Splice & Loss of function, often
- SPTBN4 protein
- Expression: Brain, Nerve, Pancreas, Skeletal muscle sarcolemma
- May modulate signal pathways & membrane excitability
- β-IV spectrin
- Enriched at axon initial segments & Nodes of Ranvier
- Forms a periodic lattice with actin
- Functions with AnkG to cluster Na+, KCNQ2 & KCNQ3-subunit-containing K+ channels
- NMJ maintenance
- Neuronal (Non-erythrocyte) spectrins
- Composition
- Subunits: α-II subunit + β-I, β-II, β-IV, or β-V
spectrin subunits
- Self association: 2 sets of subunits
- Subunits: α-II subunit + β-I, β-II, β-IV, or β-V
- Axon submembrane cytoskeleton: Repeating circumferential actin rings, connected by Spectrin tetramers
- Composition
- Spectrin disorders
- Clinical
- Onset: Neonatal to 60 years; Usual childhood
- Motor
- Hypotonia & Weakness
- Delayed development
- Respiratory & Feeding difficulties
- Weakness: Face & Distal
- Inability to sit or stand
- Muscle atrophy, diffuse
- Skeletal: High-arched palate; Scoliosis; Ankle contractures
- Tendon reflexes: Absent
- Deafness: Central
- CNS
- Mental retardation: Global; No speech
- Seizures: Some patients
- Visual impairment
- Congenital heart defects
- Laboratory
- Muscle: Denervation or Normal
- Type 1 muscle fibers: Small
- Some small esterase+ muscle fibers
- Nerve
- Axon loss, Motor
- Nodes
- Na+ channels: Reduced
- No KCNQ2 K+ channels
- NCV: 31 to 41 M/s
- Serum CK: Normal
- Brainstem-evoked potentials: Absent
- Brain MRI: Abnormal in some patients
- Muscle: Denervation or Normal
- Mouse model: Quivering
- Type 1 muscle fibers absent
- Mislocation of voltage-gated channels at axon initial segments & nodes of Ranvier
Neuromuscular disease & Ocular or Auditory anomalies ± Seizures (NMOAS)
●
DEAH Box polypeptide 16 (DHX16)
- Epidemiology: 4 patients
- Genetics
- Mutations: Gly427Glu; Phe582Ile; Gln697His; Thr674Met
- DHX16 protein
- Helicase domain-containing protein
- DExD/H-box RNA helicase family
- ATP-dependent unwinding of RNA secondary structures
- Required for pre-mRNA splicing after formation of precatalytic spliceosome
- DExD/H-box RNA helicase disorders
- Clinical
- Onset age: Congenital
- Hypotonia
- Developmental delay
- Retina pigmentary pathology
- Seizures
- Deafness
- Skeletal: Short limbs, Growth delay, Contractures in some patients
- Course: Death in infancy in 2 patients
- Laboratory
- Brain MRI: Corpus Calloum disorders
- EMG: Myopathic
- NCV: Axon loss
- Muscle: Type 1 fibers small; Minor inflammation
- EEG: Seizures
OTHER CONGENITAL WEAKNESS
Congenital Myotonic Dystrophy● Myotonin protein kinase; Chromosome 19; Dominant
- Largest # of triplet repeats of any MD syndrome (> 1,000)
- Examine parents (esp mother) for evidence of disease
● Chromosome 4q35; Dominant
- Congenital FSH often presents with no clearly affected parent
- Acid Maltase
- Debranching Enzyme
- Branching Enzyme
- Carnitine Deficiency
- Mitochondrial (Cytochrome Oxidase Deficiency)
Congenital Neuropathic Syndromes
- Spinal Muscular Atrophy: 5q & X-linked
- Congenital Neuropathies: Hypomyelinating; CIDP
Broad A band disease
- Epidemiology: 2 sporadic cases
- Clinical
- Onset: Neonatal
- Hypotonia
- Developmental delay: Motor; Speech
- Progressive improvement in function
- Retinal dystrophy: One patient
- Laboratory
- Serum CK: Mildly high
- EMG: Normal
- Muscle pathology
- Broad A bands visible on plastic sections
- Misalignment of myosin thick filaments
Trilaminar myopathy
- Epidemiology: 1 sporadic case
- Clinical
- Onset: Birth
- Tone: Increased resistance to passive movement
- Contractures
- Course: Increasing spontaneous movements
- Cognition: Normal
- Laboratory
- Serum CK: High (6x to 40x)
- EMG: Normal
- Muscle pathology
- Fiber size: Varied
- Large fibers: Trilaminar appearance
- NADH: Dark inner & outer zones
- ATPase: Pale inner & outer zones
- Ultrastructure
- Central zone lacks organized myofibrils
- AChRs: Extrajunctional, between intermediate & outer zones
Zebra body myopathy 118
● α-Actin (ACTA1; Skeletal muscle)- Epidemiology: 3 sporadic cases
- Genetics
- ACTA1 mutations: Null alleles; Leu348Gln
- Allelic disorders
- Clinical
- Onset: Birth
- Hypotonia
- Weakness
- Proximal, Distal & Axial
- Symmetric or Asymmetric
- May be greater in arms
- Face & Neck: Mild
- Muscle size: Small
- Tendon reflexes: Normal
- Course: Static or Slow improvement
- Laboratory
- Serum CK: Normal or Mildly high
- EMG: Myopathic
- Muscle pathology
- Fiber size: Varied
- Internal nuclei
- Internal architecture: Irregular
- Zebra bodies
- Nemaline rods
- Cytoplasmic bodies
Weakness with early skeletal disorders
- Tel Hashomer Camptodactyly Syndrome
● Autosomal Recessive- Causes
- Aarskog�Scott syndrome: FGD1 mutations
- Clinical features
- Skeletal: Camptodactyly (flexed fingers); Club feet; Spina bifida - Cervical
- Skin: absent interphalangeal creases; Whorls 7 or more fingers; Palmar creases
- Abnormal sweat pores
- Cardiac: Mitral valve prolapse
- Myopathy: Wasting; Variable fiber size; High serum CK
- Heterozygotes: Partial skeletal (5th finger camptodactyly) & skin manifestations
- Causes
- Camera-Marugo-Cohen Syndrome
- Clinical features
- Obesity
- Mental retardation
- Skeletal: Body asymmetry; Camptodactyly; Clinodactyly
- Muscle weakness: Proximal, Legs > Arms 50%; Congenital hypotonia 50%
- Clinical features
- Shwachman�Diamond: SBDS; 7q11
- Skeletal dysplasia, Weakness, Intellectual Disability, Ovarian Dysfunction: SOX8
Williams-Beuren syndrome
●
Autosomal dominant contiguous gene syndrome; Chromosome 7q11.23● Deleted genes: Elastin
- Genetics
- Microdeletion in region of highly repetitive DNA
- 2° to recombination between misaligned repeat sequences flanking WBS region
- Clinical
- Vascular stenoses: Supravalvular aortic; Coronary; Pulmonary; Renal
- GU: Urethral stenosis; Small or solitary kidney
- ENT: Elfin faces; Hoarse voice; Vocal cord paralysis
- Gastrointestinal: Inguinal hernia; Constipation; Diverticulosis
- Metabolic: Episodic dehydration; Hypertension; Hypercalcemia
- Mental retardation: 65 IQ average
- Deficient visuo-spatial ability
- Relatively preserved verbal function
- "Cocktail party" personality
- Attention defecit
- Hypersensitivity to sound
- Joint contractures
- Motor
- Infants: Hypotonia; Delayed walking; Fatigue
- Adults: Cramps; Myalgia; Fatigue
- Muscle pathology
- Myopathy: Variable fiber size
- Some with lipid storage & Carnitine deficiency
Neonatal perifascicular myopathy 1
- Onset
- Age: Neonatal
- Motor: Hypotonia & Weakness
- Clinical
- Motor
- Hypotonia
- Weakness: Diffuse
- CNS
- Cognitive delay
- Mental retardation: Mild
- Course
- Non-progressive
- Improvement in motor skills with increasing age
- Motor
- Muscle pathology
- Muscle fibers: Perifascicular pattern of pathology
- Atrophy
- Immature, type 2C muscle fibers
- Connective tissue
- Alkaline phosphatase staining of perimysium
- Cells: Macrophages or dendritic
- Inflammation: Perivascular, near & around intermediate sized vessels
- Muscle fibers: Perifascicular pattern of pathology
Immunoneurologic Disorder (Woods-Black-Norbury Syndrome)
●
Dominant; Chromosome Xq26-qter- Males: Neonatal hypotonia; Early death
- Females: Spastic paraparesis
- Clinical
- Weakness: Progressive
- Tendon reflexes: Brisk
- Bladder dysfunction
- Night vision: reduced
- Laboratory
- IgG2 deficiency
- Clinical
Congenital absence, weakness, or hypoplasia of muscles
|
Congenital Cranial Dysinnervation (CCDD) Extraocular muscles Axenfeld-Rieger Blepharophimosis CCDD: ECEL1; 2q37 CCDD: COLA25A1; 4q25 Congenital fibrosis Duane retraction syndromes (DRS) Holt-Oram: TBX5 Möbius syndrome (MBS) Ptosis + Scoliosis HGPPS1: ROBO3; 11q24 HGPPS2: DCC; 18q21 |
Face Cardiofacial syndrome Depressor anguli oris Mastication Möbius syndrome Hands Finger extensors Holt-Oram Palmaris longis Thenar eminence Legs Peroneus tertius Psoas (CHILD) Trunk Abdominal Brachial plexus Diaphragm Pectoral Poland syndrome Prune belly Trapezius |
Extraocular muscles
- Congenital Fibrosis of Extraocular Muscles (CFEOM)
198
CFEOM1: KIF21A; 12q12
CFEOM2: PHOX2A; 11q13
CFEOM3: TUBB3; 16q24
CFEOM4 (Tukel): 21qter
CFEOM5: COL25A1; 4q25
CFEOM + Polymicrogyria: TUBB2B; 6p25
CFEOM: TUBA1A; 12q13
CFEOM: ECEL1; 2q37
CFEOM: MYF5; 12q21
Also see
Duane retraction syndromes (DRS)
- Congenital fibrosis of extraocular muscles 1 (CFEOM1)
● Kinesin family member 21A (KIF21A)
; Chromosome 12q12; Dominant & Sporadic
- Epidemiology
- > 50 patients
- Most common CFEOM cause
- Genetics
- Mutations
46
- Heterozygous
- Missense: R954W; R954Q; Other
- Stalk region of protein: Most common
- Some de novo: More in sporadic cases
- Allelic disorders
- CFEOM3B phentype
- Fetal akinesia + Arthrogryposis multiplex, Recessive 183
- Ocular dysfunction + Developmental delay
- Polyneuropathy, CNS dysfunction ± CFEOM
- Motor neuropathy, Distal, Dominant
- Mutations
46
- KIF21A protein
- Clinical
- Ptosis: Congenital; Bilateral
- EOM
- Oculomotor nerve, Superior division: Absent
- Common defect: Inability to elevate eye
- Other: Little horizontal movement
- Nystagmus
- Eyes fixed 20° to 30° below horizontal
- Head tilt: Posterior
- Blepharophimosis
- Pathology
- Developmental absence of the superior division of oculomotor nerve
- Reduced # of midbrain α oculomotor neurons
- Anomalous extraocular muscle insertion
- Muscle changes: Superior rectus & Levator palpebrae superioris
- KIF21A variant: Polyneuropathy, CNS dysfunction ± CFEOM
197
- Epidemiology: 2 patients
- Genetics
- Inheritance: Dominant/de novo
- Mutations: Missense; Leu664Pro; Leu685Pro
- Clinical
- Onset: Childhood
- Peripheral neuropathy: Progressive
- EOM: Strabismus or EOM limitation
- CNS: Developmental delay; Motor, Speech or Intellectual
- Laboratory
- Brain imaging: Corpus callosum hypoplasia ± other White matter Δ
- KIF21A variant: Motor Neuropathy, Distal, Dominant
207
- Epidemiology: 2 patients
- Genetics
- Inheritance: Dominant or de novo
- Mutation: Leu664Pro; Heterozygous
- Clinical
- Onset age: Early Childhood
- Motor delay
- Weakness: Distal, Legs & Arms; Neck
- Muscle atrophy: Distal; Legs & Arms
- Tendon reflexes: Reduced or Absent
- Eyes: Exotropia
- Vomiting: Frequent
- Contractures: Distal
- Laboratory
- Serum CK: Midly high
- NCV: Axonal motor neuropathy; CMAPs small, SNAPs normal
- EMG: Chronic distal denervation
- Muscle MRI: Fatty replacement, diffuse in distal legs
- Epidemiology
- Congenital fibrosis of extraocular muscles 2 (CFEOM2)
● Aristaless homeobox, drosophila, homolog of (ARIX; PHOX2A); Chromosome 11q13.4; Recessive
- Mutations: Splice sites & Missense
- ARIX protein
- Homeodomain transcription factor
- Required for cranial nerve III & IV development
- Epidemiology: Saudi Arabia (Ala72Val); Turkey
- Clinical
- Limited extraocular movements
- Eyes fixed in abduction: Large angle exotropia
- Deficient function of muscles innerved by 3rd or 4th cranial nerves
- Can raise eyes above horizontal or have unilateral involvement
- Ptosis
- Visual acuity
- Poor
- Severe amblyopia: In eyes with pupil covered by ptotic lid
- Pupils
- Miotic
- Fixed or sluggishly reactive to light or pupillary dilators
- Limited extraocular movements
- Pathology
- Complete loss of oculomotor nerves for both eyes
- Congenital fibrosis of extraocular muscles 3 (CFEOM3)
● Tubulin, Beta-3 (TUBB3); Chromosome 16q24.3; Dominant
- Genetics
- TUBB3 mutations
- Missense
- R62Q; Arg262Cys (Common); R262H; Ala302Thr; R380C; Glu410Lys; Asp417His; Asp417Asn
- Locations: Regions of β-tubulin regulating microtubule dynamics, motor protein trafficking & interactions with MAPs
- E410, D417 & R262: Loss of Kinesin Localization on Microtubule Plus Ends
- A302T, R62Q & R380C: Microtubules more stable & longer lifetimes
- R262C, R262H & E410K: Duration of individual growth events prolonged; Increased rate of depolymerization
- Allelic disorders
- CFEOM + Hypopgonadism
- Sensory-Motor polyneuropathy
- Cortical dysplasia, complex, with other brain malformations 1
: 1 patient with unilateral hypohidrosis
- CFEOM3B syndrome
: Some KIF21A mutations
- TUBB3 mutations
- TUBB3 protein
- Function: Role in axon growth guidance & maintenance
- May interact with KIF21A
- See: Axonal transport
- Mutations: May impair tubulin heterodimer formation
- Clinical
- Variable expression
- Often asymmetric
- Ptosis
- Extraocular movements: Limited
- Deficient function of muscles innerved by the third or fourth cranial nerves
- Can raise eyes above horizontal or have unilateral involvement
- Mild: Limited vertical gaze
- Severe: 1° gaze fixed in hypo- & exotropic position; Marked restriction of EOM
- Marcus-Gunn: Ptotic eyelid elevation associated with synkinetic jaw movements
- Other features: Some mutations
- Aberrant innervation of cranial musculature by trigeminal nerve
- Intellectual & behavioral impairments: A302T, E410K, R262H & R380C
- Facial paralysis (Möbius)
- Axonal sensorimotor polyneuropathy: Later-onset; E410K D417H or R262H mutations
- Onset age: 1st to 3rd decade
- Leg weakness
- Sensory loss
- EMG: Chronic, generalized sensorimotor polyneuropathy
- May occur without CFEOM in some patients: CMT 2-like syndrome
- Wrist & finger contractures: D417H or R262H mutations
- Course: Non-progressive
- MRI
- Hypoplasia: Oculomotor nerve
- Hypoplasia: muscles innervated by III nerve
- No polymicrogyria
- Superior division: Levator palpebrae superioris; Superior rectus
- Inferior division: Medial rectus
- Oculomotor nerve aberrant innervation: Lateral rectus muscle
- Dysgenesis of corpus callosum, anterior commissure (AC) & internal capsule
- More severe: A302T, E410K, R262H & R380C
- Less severe: R62Q, R262C, or D417N
- White matter: Generalized loss
- Basal ganglia: Dysmorphisms correlating with specific mutations
- Cortex: Normal
- Genetics
- Variant syndrome: CFEOM3 + Hypogonadism
- TUBB3 Mutation: E410K
- Clinical
- Moebius syndrome
- Congenital fibrosis of extraocular muscles
- Facial weakness
- Polyneuropathy: Sensory-Motor; Progressive
- Kallmann syndrome (Hypogonadotropic hypogonadism & Anosmia)
- Midface hypoplasia
- Intellectual disabilities
- Some patients: Vocal cord paralysis, Tracheomalacia, Cyclic vomiting
- Moebius syndrome
- MRI
- Thin corpus callosum & anterior commissure
- Olfactory sulci & bulbs: Absent
- Oculomotor & Facial nerves: Absent
- TUBB3 variant syndrome: Polyneuropathy
- TUBB3 mutation: D417N
- Clinical
- Onset age: 2nd decade
- Polyneuropathy
- Sensory-Motor
- Gait disorder
- Progressive to wheelchair: 4th decade
- EOM: Normal
- Intellect: Normal
- Laboratory
- Axon loss: Sensory + Motor
- Congenital fibrosis of extraocular muscles 1 (CFEOM1)
- Congenital fibrosis of extraocular muscles (CFEOM) + Polymicrogyria (CDCBM7)
103
● Tubulin, Beta-2B (TUBB2B); Chromosome 6p25.2; Dominant
- Epidemiology
- Australian-based Caucasian family
- Rare cause of CF-EOM
- Genetics
- TUBB2B mutation: E421K; Heterozygous
- Alters microtubule dynamics: Increased polymer stability; Reduced TUBB2B αβ-heterodimer production
- Can reduce kinesin localization
- Other TUBB2B mutations: Polymicrogyria, symmetric or asymmetric
- TUBB2B mutation: E421K; Heterozygous
- TUBB2B protein
- Forms α/β tubulin heterodimers
- Required for neuronal migration
- Clinical
- Congenital fibrosis of extraocular muscles (CFEOM)
- Ptosis: Bilateral
- Vertical eye movements
- Severely limited
- Jerky convergent movements on attempted elevation
- Horizontal movements: Variably restricted
- Eyes fixed in downgaze: Bilateral
- Chin-up head position: Compensatory
- Pupils: Normal
- Intellectual disability
- Head circumference: < 2nd percentile
- Congenital fibrosis of extraocular muscles (CFEOM)
- Laboratory
- MRI
- Polymicrogyria
- Commissural projection neurons: Aberrant trajectories
- Caudate: Small
- Extra-ocular muscles: Small
- Nerve conduction studies: Normal
- MRI
- Mother: Mild polymicrogyria
- Epidemiology
- CFEOM with ulnar hand anomalies (Tukel sundrome)
58
● Chromosome 21qter; Recessive
- Epidemiology: 1 Turkish family
- Clinical
- Ocular
- Ophthalmoplegia
- Muscles
- Especially inferior oblique & superior rectus
- Limited upgaze: Most common
- Levator palpebrae: Some patients
- Severe cases: Complete ophthalmoplegia
- Non-progressive
- Forced ductions: Normal
- Muscles
- Blepharoptosis: Right eye
- Head posture: Chin elevation
- EOM pathology: Fibrosis
- Ophthalmoplegia
- Hands
- Postaxial oligodactyly/oligosyndactyly
- Especially absent 5th finger
- More severe on right side
- Postaxial oligodactyly/oligosyndactyly
- Ocular
- CFEOM5: Congenital Cranial Dysinnervation Disorder (CCDD)
117
● Collagen 25A1 (COL25A1); Chromosome 4q25; Recessive
- Epidemiology: 2 families
- Genetics
- Mutations: Gly382Arg; Gly497Ter
- COL25A1 protein
- Expression: Ubiquitous; CNS; Eye muscle
- Proteolysis produces: Soluble collagen-like amyloidogenic component (sCLAC)
- sCLAC: Component of amyloid plaques
- Role in NMJ formation
- Clinical
- Onset age: Congenital
- Eye
- Ptosis: Bilateral; Severe
- Duane syndrome
- CFEOM
145
● Myogenic Factor 5 (MYF5); Chromosome 12q21.31; Recessive
- Nosology: External Ophthalmoplegia + Rib & Vertebral Anomalies (EORVA)
- Epidemiology: 3 families (Turkey & Yemen)
- Genetics
- Mutations: Loss of function; p.Gln8Leufs*86 (10 bp deletion), Arg95Cys
- MYF5 protein
- basic helix-loop-helix (bHLH) transcription factor
- Clinical
- Onset age: Congenital
- Eye: External ophthalmoplegia; Strabismus; Ptosis
- Motor: Generally normal development & tone
- Cognition: Normal
- Skeletal
- Ribs: Hypoplastic or Missing
- Spine
- Torticollis
- Scoliosis
- Vertebral anomalies: Fusions; Narrow disc spaces
- Course
- Spine anomalies: Progressive
- Other: Non-progressive
- Laboratory
- Orbital MRI: All EOM hypoplastic or Absent
|
General DURS1: 8q13 DURS2: CHN1; 2q31 DURS3: MAFB; 20q12 DRRS (Okihiro): SALL4; 20q13 BSAS: HOXA1; 7p15 Navajo: HOXA1; 7p15 DA5E: PIEZO2; 18p11 CCDD: COLA25A1; 4q25 Other DURS-like syndromes |
|

- Clinical features, Duane syndromes: General
Duane-like phenomenon in patient
diagnosed as chronic ocular myasthenia gravis.
- Mild ptosis in R eye on center gaze. (Top)
- Increased ptosis & retraction in R eye
with adduction (L gaze). (Bottom)
- Restriction of EOM
- Type 1: Limited abduction
- Most common
- Strabismus: Esotropia
- VI nerve: Absent or Hypoplastic
- Lateral rectus muscle: Aberrant innrevation by III nerve
- Type 2: Limited adduction; Least common
- Type 3: Limited abduction & adduction
- Type 1: Limited abduction
- Family history
- Sporadic: Usually unilateral
- Familial: Often bilateral
- Bilateral syndromes
- Amblyopia & Strabismus: More common
- Especially familial cases
- Eye adduction results in
- Globe retraction
- Palpebral fissue narrowing
- Associated congenital malformations common
- Skeletal
- Ear
- Ocular
- Neural
- Pathology
- Absent abducens nuclei + nerve
- Lateral rectus muscle
- Denervated
- Aberrant innervation by inferior division of oculomotor nerve
(Also innervates ipsilateral medial rectus muscle)
- DURS1
● Chromosome 8q13; ? Sporadic
- Genetic: Deletion in 8q rearranged region
- Clinical: Multisystem disorder
- EOM
- Abduction: Marked or Complete limitation
- Adduction: No limitation
- Retraction & Ptosis: With adduction
- Skeletal: C2-C3 vertebral fusion (Klippel-Feil)
- Thenar hypoplasia
- Deafness
- EOM
- DURS2
72
● α2-Chimerin (CHN1); Chromosome 2q31; Dominant
- Epidemiology: Mexican & UK families
- Genetics: Mutations
- Missense: Heterozygous
- Gain-of-function: Increase α2-chimaerin RacGAP activity
- Some mutations enhance
- α2-chimaerin translocation to cell membrane
- Ability of α2-chimaerin to self-associate
- Functional effect: Axonal disorder
- Aberrant branching and/or defasciculation of nerve
- CHN1 protein
- Rac-specific GTPase-activating: Down-regulates Rac activity
- Binds to diacylglycerol (DAG): Membrane associated phorbol ester signaling lipid
- Strongest expression during development in midbrain & hindbrain
- Congenital ophthalmoplegia
- Abduction: Absent or Reduced
- Adduction: Reduced in some patients
- Eye retraction & ptosis: On adduction
- Higher incidence of
- Bilateral involvement
- Vertical movement abnormalities
- Pathology
- Absent abducens nerve & nucleus in pons
- Hypoplastic oculomotor nerves: Small oculomotor-innervated muscles
- Mice with loss of α2-chimaerin
- Disrupted ephrin/EphA4 signaling
- Elevated RacGTP levels
- Phenotype: Hopping rabbit-like gait resulting from
- Pathology: Excessive & aberrant midline crossing of
- Corticospinal tract axons
- Spinal interneuron projections
- Duane anomaly + Radial ray abnormalities & Deafness (DRRS; Okihiro syndrome)
● Sal-like 4 (SALL4); Chromosome 20q13.2; Dominant
- Allelic with: IVIC syndrome
- Clinical
- Upper limb: Hypoplasia of thenar muscles; Absence of thumb & first metacarpal
- Ocular: Duane syndrome; Coloboma
- Renal: Malrotation; Absence
- Ear: Deafness; Narrow external auditory meatus
- Differential diagnosis: Holt-Oram syndrome
- Allelic with: IVIC syndrome
- Duane Retraction Syndrome 3 (DURS3)
130
● V-MAF Musculoaponeurotic Fibrosarcoma oncogene family, protein B (MAFB);
Chromosome 20q12; Dominant
- Epidemiology: 4 families; 1% of DRS probands
- Genetics
- Mutations: Deletion of 1 base pair or whole MAFB gene
- Loss of function
- MAFB protein
- Location: Nucleus
- DNA binding
- Interacts with intracellular domain (ICD) of Lrp1
- Expressed in rhombomeres 5 & 6, where VI nerve develops
- Clinical
- Duane syndrome
- Abduction: Bilateral or Unilateral limitation
- Adducted eye: Ptosis; Retraction
- Type 1 or 3 pattern
- Deafness
- 1 family
- Inner-ear common-cavity anomalies
- Bosley-Salih-Alorainey Syndrome (BSAS)
● Homeobox A1 (HOXA1);
Chromosome 7p15.2; Recessive
- Epidemiology: Saudi-Arabian, Turkish & Native American (Navajo) families
- Genetics
- Mutations
- Stop: 175-176insG, R26X, Y28X
- Homozygous
- Mutations in other HOX genes: Cause malformed distal extremities
- Mutations
- Clinical
- Duane syndrome: Bilateral
- Horizontal gaze: Abduction & Adduction limited
- Up & down gaze: Preserved
- Convergence: Normal; Associated globe retraction
- Deafness & Mutism (89%)
- Motor development (66%): Delayed; Walking > 3 years
- Other CNS (22%)
- Mental retardation
- Autism spectrum disorder
- Duane syndrome: Bilateral
- Malformations
- Carotid
- Hypoplasia or absence of skull canals & arteries: Internal carotid often absent
- Compensatory enlargement of basilar & posterior communicating arteries
- Ear: Absent cochlea, semicircular canals & vestibule
- VI nerve: Absent
- Normal: Cerebellum, Brainstem, Cerebrum
- Carotid
- Allelic disorder: Athabascan (Navajo) brainstem dysgenesis syndrome (ABDS)
- Mutation: R26X
- Onset: Congenital
- Clinical: BSAS +
- Face weakness
- Central hypoventilation
- Vocal cord paralysis: 20%
- CNS
- Hypoventilation
- Developmental delay: Global
- Seizures: 50%
- Heart
- Congenital defects: 50%
- Outflow tract anomalies in some
- BAERs & Caloric testing: No response
- Similar syndrome to HOXA1-/- mice
- Duane syndromes: Other multisystem or sporadic
- Karyotypic change on Chromosomes 4 or 22
- Wildervanck syndrome
- Duane anomaly
- Klippel-Feil anomaly (Fused cervical vertebrae)
- Deafness: Congenital perceptive
- Mostly females
- Bosley-Salih-Alorainey Syndrome (BSAS)
- Neurodevelopmental disorder, Dysmorphic facies & Distal skeletal anomalies (NEDDFSA)
● ZMIZ1;
Chromosome 10q22.3; Recessive
|
|
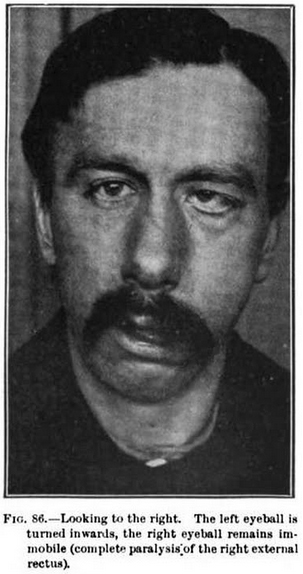
|
 Bramwell 1907 |
● ROBO3
- Epidemiology: 10 families, Turkish, Indian, Pakistan, Greek, Arab
- Genetics
- Mutations: Missense; 1 bp insertion; Splice site
- ROBO3 protein
- Expressed by commissural axons in brainstem
- ? Allows axons to cross the midline
- Clinical
- Onset: 1st decade or Congenital
- Gaze paresis
- Horizontal: No conjugate horizontal smooth pursuit, saccades, or vestibuloocular reflex
- Vertical: Spared
- ? Supranuclear
- Esotropia: 30%
- Nystagmus: Pendular
- Facial myokymia (1 patient)
- Scoliosis: Mild to Moderate
- Mental retardation: 30%
- No weakness
- MRI
- Brainstem: Dysplastic in some patients
 Bramwell 1907 |
● Deleted in colorectal carcinoma (DCC)
- Epidemiology: Mexican & Saudi families
- Genetics
- Mutations: Stop; Intragenic deletions
- Allelic disorders
- Colorectal cancer, somatic
- Esophageal carcinoma, somatic
- Mirror movements 1
- Corpus callosum agenesis: Missense (Q691K) mutation
- Colorectal cancer, somatic
- DCC protein
- Transmembrane protein
- Immunoglobulin superfamily
- Expressed on spinal commissural axons
- Possesses netrin-1-binding activity
- Antibody target: With serum CASPR2 antibody & Thymoma
- Clinical
- Developmental delay: Global
- Hypotonia
- Walking: Delayed
- Intellectual disability
- Mirror movements: Males
- Gaze palsy: Horizontal
- Scoliosis: Childhood-onset; Progressive
- Developmental delay: Global
- Laboratory
- Brain imaging
- Corpus callosum: Agenesis
- Anterior & hippocampal commissures: Absent
- Pons & Midbrain: Hypoplasia; Midline cleft
- Brain imaging
- External link: DJO
● Dominant
- Eyes
- Hypoplastic anterior iris
- Absent eye muscles, partial
- Proptosis
- Hypertelorism
- Ears: Sensorineural deafness, mild
- CNS
- Communicating hydrocephalus
- Psychomotor retardation
- Short stature
● Forkhead box K2 (FOXK2; ILF1)
- Epidemiology: 5 families; 1 with 4 generations
- Genetics
- Mutations: Missense; R215W, T55I, R215W, P411A, R544Q, Q550H
- FOXK2 protein
- Transcription factor
- DNA-binding forkhead box domain (FOX)
- Nuclear localization sequence
- Regulator of metabolic reprogramming towards aerobic glycolysis
- Autophagy: Negative regulator in skeletal muscle
- Mitochondrial metabolism
- Regulation in adipocytes
- Affects expression of mitochondrial function related genes: Pdk4
; Cry1
- FOXK2 deficiency
- Skeletal muscle development & impaired motor ability: Abnormal in zebrafish
- Mitochondrial homeostasis: Abnormal
- Coenzyme Q10: May alleviate disorders
- Clinical
- Onset: Congenital
- Ptosis: Isolated
- Laboratory
- Eye muscle: Abnormal mitochondrial morphology
● Type 1
- Ptosis: Symmetric or Asymmetric
- Normal eye movements
- Penetrance: Incomplete
● Type 2
- Male = Female
- Penetrance: High
- Onset: Congenital
- Bilateral: Symmetric
- Normal eye movements
-
Retardation of growth & development
- Clinical
- Retardation: Growth; Mental
- Face: Ptosis, Epicanthus, Broad nasal bridge, Low-set & abnormal ears
- Congenital heart defect
- Anal atresia
Ocular ptosis
Cardiac defect
Anal atresia
- Definition: Palpebral fissure has fixed reduction in vertical dimension

- Blepharophimosis 1
:
+ Ptosis & Epicanthus inversus
● FOXL2;
Chromosome 3q22.3; Dominant
- Genetics
- Polyalanine repeat expansions (30%): May cause blepharophimosis syndrome without ovarian failure
- Clinical
- Ptosis
- Telecanthus
- Lid phimosis
- Epicanthus inversus: Small skin fold arising from lower lid running inwards and upwards toward nose
- ± Ovarian failure
- Genetics
- Saethre-Chotzen Syndrome (SCS; Blepharophimosis 2)
● TWIST
Chromosome 7p21.1; Dominant
- Blepharophimosis with short stature
● Autosomal recessive- Family: Yeminite-Jewish
- Clinical
- Eyes: Blepharophimosis; Ptosis; Ophthalmoplegia
- Face: Prognathism; Synophrys; Thick eyebrows; Weak frontalis
- Skeletal: Short stature; Syndactyty toes 2 & 3
- CNS: Borderline mental retardation; Anosmia
- Ohdo syndromes
- Genetics: Several sub-types
- Deletions of chromosome 3 short arm
- Ohdo type: Typical clinical features, No hypotonia
- Verloes type: Brain malformation & Epilepsy
- Say/Barber/Biesecker/Young/Simpson (SBBYS) type
: KAT6B mutations
; Hypotonia, Hyperextensible joints
- Maat-Kievit-Brunner (MKB) type
: MED12 mutations
; Thick alae nasi, Triangular face
- Clinical: General features
- Mental retardation
- Congenital heart disease
- Eyes: Blepharophimosis; Blepharoptosis
- Teeth: Hypoplastic
- Genetics: Several sub-types
- Schwartz-Jampel syndrome
- Dubowitz syndrome
- Ptosis
- Blepharophimosis
- Lateral telecanthus
- Skeletal
- Microcephaly
- Short stature
- Mental retardation (50%)
- Marden-Walker syndrome
Face
Depressor anguli oris: Cardiofacial syndrome● Chromosome 22q11.2 (deletion) Dominant; Also Multifactorial
- Nosology: Cayler syndrome; Di George syndrome
- Genetics: Often paternally derived chromosome
- Facial weakness: Variable severity
- Congenital
- Mild: Unilateral lower lip
- Severe: Complete facial paresis
- Cardiac
- Atrial septal defect
- Ventricular septal defect
- Myopathy
20
- Strength: Normal
- Serum CK: May be high
- Muscle biopsy: Variation in fiber size; Increased glycogen
- Skeletal
- Cervical vertebral fusion
- Adducted thumbs
- Facial dysmorphism
- Bifid uvula
- Immune
- T-cell abnormalities
- Thymic hypoplasia
- CNS (10%)
- Microcephaly: Variable
- Mental retardation
Also see: Congenital facial paresis
Absent Muscles of Mastication: Mouse model
- Double knockout of MyoR (Musculin)
and capsulin (TCF21)
- Phenotype
- Absent muscles in 1st branchial arch: Masseter; Pterygoids, Medial & Lateral; Temporalis
- Cleft palate
- Diaphragmatic hernia: Posterior
Diaphragm: Weakness & Defects 78
- Diaphragm function
158
- Contraction generates: Negative intrathoracic & positive abdominal pressure
- Inspiratory pump: 80% of power of respiration
- Partition between thoracic & abdominal cavities: Muscularized Only in mammals
- Active 60% of time: vs 2% to 15% in limb muscles
- Phrenic nerve: General
- Chief muscle of inspiration
- Motor innervation
- Roots: C3, C4, C5
- Phrenic nerve
- Sensory innervation
- Phrenic nerve
- Intercostal nerves
- Lesions
- Infants: More severely clinically affected
- Clinical: Unilateral
- 50% Asymptomatic
- 25% Exertional dyspnea, mild
- Vital capacity: 75% of predicted
- Clinical: Bilateral
- Exertional dyspnea: Severe
- Orthopnea
- Nocturnal hypoxemia & hypercapnia
- Vital capacity: 45% of predicted; Supine < 75% of upright
- Radiology: Elevated diaphragm on afffected side
- Diaphragm Disorders: Hereditary
- General
- Bilateral & left side reported
- Usually recessive
- General types of congenital diaphragmatic hernia (CDH)
- Bochdalek
- 70 to 90% of CDH cases
- Left sided
- Posterolateral
- Morgagni: Anterior retrosternal
- Central CDH
- Midline of septum transversum
- 1 to 2% of CDH
- Congenital (DIH)
- X-linked
- Bochdalek
- Specific syndromes
- Fryns
: Coarse facies; Cloudy corneae, Diaphragm defects, Lung lobulation absent; Distal limb deformity
- Tonne-Kalscheuer
: RLIM
; X-linked
- Simpson-Golabi-Behmel, type 1 (SGBS1)
: GPC3
; X-linked, Rec
- Pallister-Killian syndrome (Tetrasomy 12p; PKS)
- Cornelia de Lange syndrome (CDLS1)
: NIPBL
; Dominant
- Lethal multiple pterygium
- MYODRIF: MYOD1
- SMARD1 (HMN 6; DSMA1)
- EMARDD (Early onset myopathy, areflexia, respiratory distress & dysphagia)
- MDC1B
- Myotubularin female carriers: Elevated hemidiaphragm
- Acid maltase deficiency
- SMARD
- Fryns
- Diaphragmatic hernia, Congenital (DIH)
- DIH1
: Chromosome 15q26.1
- DIH2
: Chromosome 8p23.1
- Frequently unilateral
- Associations: Hypoplasia of left lung; Atrial septal defect
- DIH3
:
Zinc finger protein, multitype 2 (ZFPM2)
; Chromosome 8q23.1
- DIH4 + Cardiovascular defects
:
Aldehyde dehydrogenase 1 family, Member A2 (ALDH1A2)
; Chromosome 15q21.3
- Diaphragmatic defects with skull ossification & limb deficiencies
: ? Autosomal recessive
- Congenital Anterior Diaphragmatic Hernia
- ? X-linked vs. multifactorial with high male:female sex ratio
- Frequent neonatal death
- DIH1
- General
- Acquired
- Traumatic
- Etiologies
- Surgery
- Open-heart surgery
- Lung transplantation
- Esophageal surgery
- Mediastinal procedures
- Neck or thorax trauma
- Surgery
- Associated with brachial plexus injury
- Course: Biphasic worsening
- 1st hours: Tachypnea & O2 requirement
- Days to weeks: Atelectasis or infection
- Diagnosis: Chest fluoroscopy
- Prognosis
- Recovery: 50%; Onset in 1st 2 weeks
- Mortality: 10% for unilateral & 50% for bilateral
- Treatment: Plication of affected diaphragm
- Etiologies
- Phrenic nerve lesions: Other
- Neoplasm
- Herpes zoster
77
- Unilateral
- ALS
- NMJ: Myasthenia gravis
- Muscle disorders
- Chronic obstructive pulmonary disease (COPD)
- Muscle fiber atrophy
- Fiber changes: More type I; Myosin loss; Sarcomere disruption
- Amyloid
- Chronic obstructive pulmonary disease (COPD)
- Also see: Respiratory failure
- Traumatic
CMYO17: Congenital Myopathy with Diaphragmatic defects, Respiratory Insufficiency, dysmorphic Facies
● Myogenic differentiation antigen 1 (MYOD1; MYF3))
- Nosology: MYODRIF; CMYP17; CMYO17
- Epidemiology: 4 families; 6 patients
- Genetics
- Mutations: Stop
- MYOD1 protein
- Tissue expression: Skeletal muscle only
- Transcription factor: Myogenic differentiation & repair
- Clinical
- Prenatal: Polyhydramnios
- Onset age: Birth to 4th decade
- Respiratory: Insufficiency; Diaphragm high dome or hernia; Lung hypoplasia
- Motor delay
- Weakness: Diffuse or Proximal
- Skeletal: Growth, reduced; Pectus excavatum; Dysmorphic face; Distal arthrogryposis; Clinodactyly
- Renal anomalies: Some patients
- Severity: Wide variation
- Cognition: Normal
- Laboratory
- Chest XR: Diaphragm dome high
- Serum CK: Normal
- Muscle: Myopathy
Hands
Holt-Oram Syndrome● Human transcription factor TBX5
- T-Box 5 protein: Transcription factor
- Clinical
- Cardiac: ASD; VSD; AV block
- Skeletal
- Bilateral; Asymmetric, Left > Right in 70%
- Thumb (85%): Absence; Hypoplasia; Triphalangeal
- Radius (64%): Absence or hypoplasia
- Contractures: Elbow; Wrist
- Severe cases: Phocomelia
- Absent or hypoplastic muscles
- Course: Non-progressive
- Thenar (100%): Non-opposing thumb
- Trapezius: Scapular winging; Sloping shoulders
- Other: Deltoid; Biceps; Triceps; Wrist Extensor; Ulnar abductors; Supinator; Scapular winging
- Lower limbs: Normal
- TBX5 variant syndrome: Motor neuropathy, Demyelinating
- Epidemiology: 1 patient
- Genetics
- Mutation: Asp111Tyr
- Inheritance: Dominant
Palmaris longus muscle
● Autosomal dominant with incomplete penetrance
- Frequency: 12%
- Unilateral or Bilateral
- More frequent: Females (1.5:1); Left side
● Autosomal Recessive
- Unilateral finger extensor hypoplasia
- Flexion contracture
- Polyneuropathy: Sensory with hypohydrosis
- Also see: Holt-Oram
Trunk
|
Pectoral (Poland syndrome)
● Usually sporadic
|
|
||||||||
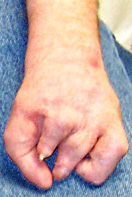
Trapezius
● Chromosome 8q12.2-q21.2; Dominant
Hypoplasia of Shoulder & Neck muscles 126
● Chromodomain helicase DNA-binding protein 7 (CHD7)
- Epidemiology: 1 family; 3 patients
- Genetics
- Mutation: Missense; Thr1133Met; de novo
- Allelic disorders
- CHARGE syndrome
- Hypogonadotropic hypogonadism 5 ± Anosmia
- Contiguous gene syndrome
- CHARGE syndrome
- CHD7 protein
- Transcriptional regulator
- Binds to enhancer elements in nucleoplasm.
- Positive regulator of ribosomal RNA (rRNA) biogenesis in nucleolus
- Development of head & neck structures
- Clinical
- Onset age: Early childhood
- Hypoplastic muscles: Trapezius; Sternocleidomastoid
- Scapular winging
- Weakness: Proximal
- Shoulders: Sloping, asymmetric; Anteverted
- Semicircular canal defects
- Dysmorphic: Small ears; Hockey stick palmar creases
- Laboratory
- Serum CK: Normal
- Muscle pathology: Non-specific
- EMG: Chronic denervation
- Muscle MRI: Trapezius bulk reduced, asymmetric
- Semicircular canals: Superior semicircular aplasia bilateral; Horizontal dysplastic
● Autosomal Dominant
- Entrapment of suprascapular nerve: Adult onset
- Also see: Erb's palsy
Abdominal musculature: Prune belly syndrome
● Cholinergic receptor, muscarinic, 3 (CHRM3)
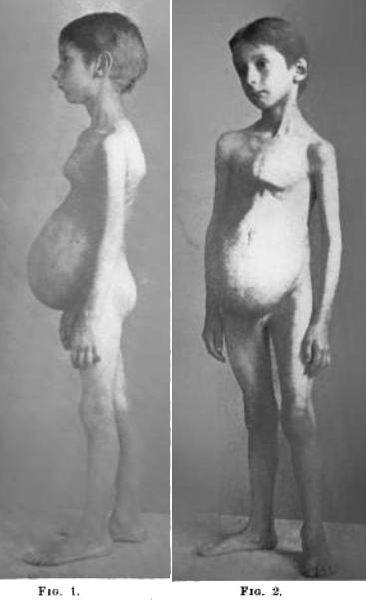 Osler 1901 |
- Nosology: Prune belly; Eagle-Barrett syndrome
- Epidemiology: 1 Turkish family
- Genetics
- Mutations
- Type: Deletion/Insertion; Frameshift; p.Pro392Alafs*43
- Homozygous
- Similar syndrome in CHRM3 null mice
- Mutations
- CHRM3
- Major receptor mediating urinary bladder contraction with micturition
- Clinical
- General: Full syndrome more common in males
- Onset age: Congenital
- Absent abdominal muscles: Abdominal distension
- Autonomic
- Pupil constriction to light: Impaired
- Dry mouth
- GU
- Urinary obstruction: Bladder outflow obstruction (BOO)
- Cryptorchidism
- Imperforate anus
- Skeletal: Pectus excavatum; Club foot; Hip dislocation
- Cardiac: Patent ductus arteriosus
- Laboratory
- Cystogram: Massive & irregular-walled bladder with diverticulae
- Variant: Similar syndrome with no CHRM3 mutation in Adult
88
- Clinical
- Onset age: Childhood
- Micturition failure: Dilated urinary bladder; Since childhood
- Pupils
- Mydriasis
- No constriction with light, near vision or muscarinic stimulation
- Hypohidrosis
- Laboratory
- No M3 protein in bladder tissue
- Differential diagnosis
- Clinical
Legs
Peroneus tertius muscle● Autosomal recessive
- Peroneus tertius muscle
- Present in man & horses
- Innervation: Deep peroneal nerve
- Action: Dorsiflexion & eversion of foot during swing phase of gait
- Levels foot & helps toes to clear ground
- Improves economy of bipedal walking
- Absence of muscle
- Frequency: 4.4% to 15% of limbs
- More frequent on right side (14:9)
Psoas (CHILD Syndrome)
● NAD(P)H Steroid dehydrogenase-like protein
● Emopamil-binding protein
- Congenital Hemidysplasia with Ichthyosiform erythroderma and Limb Defects
- Hypoplasia
- Systemic: Lung; Thyroid
- Psoas muscle
- Cranial nerves V, VII, VIII, IX & X
- CNS: Pons; Medulla; Cerebellum; Spinal cord
Multicore (Minicore) Myopathy
12
|
General Pathology ACADS: 12q24 ACTN2: 1q42 Arthrogryposis CCDC78: 16p13 Hypogonadism MEGF10: 5q23 MYH2: 17p13 MYH7: 14q11 Ophthalmoplegia RYR1: 19q13 SECISBP2: 9q22 Selenon: 1p36 Titin: 2q31 UNC45B: 17q12 Cores |
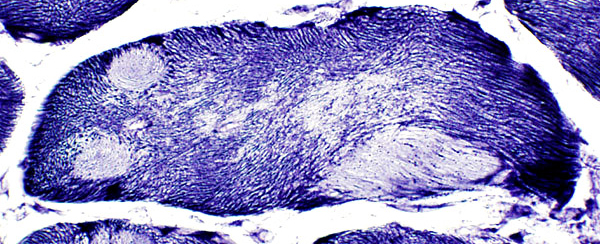 Multicores
|
Multicore myopathy: General Clinical Features
- Genetics
- Onset: Varied
- Age: Anti-natal to Childhood
- Delayed motor milestones
- Four general presentations
- Classic
- Weakness: Axial predominance
- Scoliosis: Severe
- Respiratory insufficiency
- Mild
- Weakness: Generalized; Especially pelvic girdle
- Hands: Weakness; Amyotrophy; Hyperlaxity
- Ophthalmoplegia
- Antenatal: Arthrogryposis
- Classic
- Features usually normal
- Serum creatine kinase levels
- Intellect
- Cardiac
- No malignant hyperthermia
- Muscle pathology
Multicore (Minicore) Disease
● Selenoprotein N, 1 (Selenon; SEPN1)
- General
- Common form: 30% of Mini-Multicore families
- More severe
- Genetics
- Clinical
- Early onset features
- Fetal movements: Reduced
- Hypotonia
- Weakness
- Early
- Motor delay
- All eventually ambulant
- Distribution
- Axial > Proximal > Distal
- Upper & Lower extremities
- Facial: Common; Mild
- Ocular
- Ptosis
- Ophthalmoplegia
- Some patients
- Partial to complete (10%) paresis
- Respiratory: More common with
- Age > 10 years
- Scolosis > 60°
- Nocturnal hypoventilation
- Course: Non- or Slowly progressive
- Early
- Muscle bulk: Amyotrophy
- Skeletal
- Contractures: Mild or absent
- Ligamentous laxity
- Feet: Flat or everted feet; Genu valgus
- Knees: Hyperextended
- Lumbar lourdosis
- Rigid spine
- Scoliosis: > 10 years
- Cardiac (Occasional): Septal defects; Conduction block; Cardiomyopathy
- Intelligence: Normal
- No malignant hyperthermia
- Early onset features
- Laboratory
- CK: Normal or slightly elevated (20%)
- EMG: Myopathic, especially > 4 years & Proximal muscles
- Muscle MRI: Involvement of vasti; Sparing of rectus femoris
- Pathology: Muscle
- Muscle fiber size: Variable
- Dystrophic features: Some patients
- Regenerating muscle fibers
- Endomysial connective tissue: Increased
- Multiple small cores in individual muscle fibers
- Histochemistry: Reduced activity on NADH & Mitochonrial stains
- Short: Do not extend whole length of muscle fiber
- Frequency: Present in 60% to 90% of muscle fibers
- Occur in both type I & II muscle fibers
- More prominent with greater age
- Ultrastructure: Unstructured central regions; Disordered sarcomere
- Type I muscle fibers
- Predominance
- Occasional smallness
- Internal nuclei
- Cores smaller than with RYR1 mutations
Minicore myopathy with Ophthalmoplegia
- Genetics: Several associated genes
- RYR1
: Dominant or Recessive (CMYP1B)
- SEPN1
- Short-chain acyl�CoA dehydrogenase deficiency (ACADS)
- RYR1
- Clinical
- Onset: Congenital or 1st year
- Motor
- Hypotonia
- Feeding difficulties
- Axial & shoulder girdle involvement
- Respiratory function: Reduced; Occasional failure
- Muscle MRI: More involvement of sartorius & soleus
- Weight: Low
- Scoliosis
Minicore myopathy: Antenatal onset with Arthrogryposis
- Inheritance: Recessive
- Clinical
- Arthrogryposis, generalized
- Head
- Dolichocephaly
- Prominent nasal root
- Oblique palpebral fissures
- High arched palate
- Low-set ears
- Short neck: With mild pterygium colli
- Spine: Kyphosis or kyphoscoliosis, Early & severe
- Other features
- Thorax, bell-shaped
- Clinodactyly,
- Cryptorchidism, bilateral
- Single palmar crease
- Weakness
- Proximal
- Mild
- Vital capacity reduced
Multicore myopathy: Mental retardation, Short stature & Hypogonadotropic hypogonadism (Chudley syndrome)
- Inheritance: Recessive
- Clinical
- CNS: Mental retardation
- Skeletal
- Short stature
- Lordosis: Lumbar
- Sexual infantilism
- Weakness: Generalized; Face
- Ptosis, bilateral
- Laboratory
- Pituitary fossa: Small
- Hypogonadotropic hypogonadism
- Muscle
- Fiber diameter: Varied
- Type I muscle fibers: Small
- Internal nuclei
- Multicores
CMYO5: Multicore disease, Congenital Myopathy + Fatal Dilated Cardiomyopathy
● Titin
- Nosology: Salih myopathy (SALMY); EOMFC; CMYP5; CMYO5
- Epidemiology: Moroccan & Sudanese families
- Genetics
- Titin mutations
- Homozygous or Compound heterozygous
- Inheritance: Recessive
- Allelic disorders
- Mutation carriers: Normal
- Titin mutations
- Titin protein
- Clinical
- Delayed motor milestones
- Hypotonia at birth: Some patients
- Weakness
- Onset: < 1 year
- Symmetric
- Generalized: Proximal + Distal + Face
- Legs > Arms
- Ptosis: Some asymmetric
- Course: Slowly progressive; Ambulation present in 2nd decade
- Muscle size
- Upper limbs: Atrophy
- Legs: Pseudohypertrophy, especially thighs & calves
- Contractures
- Spinal rigidity
- Joint & Neck
- Cardiomyopathy
- Progressive
- Dilated
- Onset ages: 5 to 12 years
- Rhythm disturbances: Ventricular
- Interstitial fibrosis
- Sudden death
- Laboratory
Congenital myopathy with Internal nuclei & Atypical Cores (Centronuclear myopathy 4 (CNM4)
● Coiled-coil domain-containing protein 78 (CCDC78)
- Epidemiology: 1 family
- Genetics
- Mutation: Splice-acceptor variant; Causes 222 bp inframe insertion
- CCDC78 protein
- Expressed in skeletal muscle
- Enriched in perinuclear regions & triad
- May co-localize with DHPR1α
- Clinical
- Onset
- Age: Congenital
- Hypotonia
- Weakness
- Distal > Proximal, or Diffuse
- Ambulation preserved
- Respiratory, Face & EOM: Normal
- Progression: Mild or none
- Fatigue
- Myalgias
- CNS: Mild cognitive impairment
- Onset
- Laboratory
- Muscle pathology: Centronuclear + Core
- Internal nuclei: Single; Often central; 10% to 25% of fibers
- Type I fiber predominance
- SDH: Cores, atypical & small clear regions; Irregular dark regions
- Muscle fiber aggregates: Contain CCDC78; Desmin; Actin (around clear center)
- Serum CK: Normal or Slightly high
- EMG: Normal
- Muscle pathology: Centronuclear + Core
CMYO8: Multiple structured Cores & Z-line Abnormalities
● Actinin, α-2 (ACTN2)
- Nosology: MsCD; MYOCOZ; CMYP8; CMYO8
- Epidemiology: 5 families
- Genetics
- Mutations
- Missense Asn480Ser (Recessive); Cys487Arg; c.2180T>G (p.Leu727Arg)
- In-frame deletion c.2194_2226del (p.(Ala732_Ile742del)
- Allelic disorders
- Mutations
- ACTN2 protein: Z-line
- Clinical
- Onset age: Infancy to 6th decade
- Younger onset: Hypotonia; Ophthalmoparesis
- Weakness
- Distribution: Axial, Proximal & Distal; Respiratory; Bulbar; Face
- Symmetric
- Muscle size: Atrophy
- Tendon reflexes: Reduced
- Contractures
- Cardiac: Some patients
- Course: Slow progression
- Laboratory
- Serum CK: Normal
- Muscle
- Internal architecture
- Cores: Small; Structured
- Multicores
- Motheaten fibers
- Rods
- Z-lines: Jagged
- Fiber sizes: Varied
- Internal nuclei
- Fiber types: Type 1 predominance
- Endomysial connective tissue: Increased
- Internal architecture
- Muscle MRI: Tongue; Soleus; Medial gastrocnemius; Proximal muscles (Posterior thigh)
- ACTN2 variant syndrome: Distal myopathy 6 (MPD6)
151
- Epidemiology: 6 families
- Genetics
- Inheritance: Dominant
- Mutations
- Missense; ACTN2 c.1459T>C (p.C487R); c.392T>C (p.L131P)
- Deletion in last exon: c.2558del; c.2567del 193
- Clinical
- Onset age: 34 to 63 years; Mean 40 years
- Weakness
- Early: Distal legs, especially ankle dorsiflexion
- Asymmetric
- Progression: To posterior leg & thigh muscles; Distal arms
- Face: With deletion mutations
- Myalgia (30%)
- Muscle size
- Early: Hypertrophy (Quadriceps)
- Late: Atrophy
- Cardiac: Few patients involved
- Laboratory
- EMG: Myopathic
- Serum CK: Normal to 5,000; High earlier in disease course
- Muscle MRI: Anterior leg early; Progression to thigh & glutei
- Muscle pathology
- Fiber size: Varied, Myopathic groups
- Endomysial connective tissue: Increased
- Vacuoles, rimmed
- Inclusion bodies & Aggregates: Eosinophilic; Congophilic; p62+
- Fiber morphology: Lobulation; Central lesions, core-like
- Type I predominance
- Ultrastructure
- Myofibrillar pathology
- Sarcomeres disorganized
- Cytoplasmic body-like disorders
Congenital Multi-Minicore Myopathy (CMYO9) 155
● FMR1 autosomal homolog 1 (FXR1)- Nosology
- More severe disease: Myopathy, congenital, with Respiratory insufficiency & Bone fractures (CMYO9A; CMYP9A; MYORIBF)
- Milder disease: Congenital proximal myopathy with Minicore lesions (CMYO9B; CMYP9A; MYOPMIL)
- More severe disease: Myopathy, congenital, with Respiratory insufficiency & Bone fractures (CMYO9A; CMYP9A; MYORIBF)
- Epidemiology: 6 families, 13 patients
- Genetics
- Mutation location: 5' end of Muscle-specific exon 15
- Mutations: Stop; Nucleotide deletions
- Allelic disorders; Neuropsychiatic syndromes
- FXR1 protein (FXR1P)
- 7 isoforms
- Myoblasts: Short & medium isoforms
- Cardiac & Skeletal muscle: Larger isoforms with additional exon 15
- Disorders altering isofomrs: Myotonic dystrophy; FSH dystrophy
- RNA binding
- Expression
- Proliferating cells
- Cells
- Cytoplasm & Nucleoplasm
- Associated with microtubule elements
- Tissues: Muscle, Gonads, CNS
- Muscle: FXR1 isoform p81-84; In structures within contractile bands
- MRE11 binding
- Oxidative stress responses
- Scleroderma: Autoantigen
- 7 isoforms
- Clinical
- Onset: Prenatal & Infancy
- Prenatal: Oligohydramnios; Reduced movements
- Milestones: Delayed
- Weakness
- Early: More severe patients
- Respiratory insufficiency
- Feeding: Poor
- Hypotonia
- Milder patients
- Proximal: Limb-Girdle
- Neck flexors
- Delayed walking
- Sleep apnea
- Early: More severe patients
- Tachycardia
- Skeletal
- Bone fractures: More severe disease
- Dysmorphism: Short neck
- Limbs: Ulnar deviation of hands; Laterally deviated feet
- Joint hyperlaxity
- Cognition: Normal
- Psychiatric disorders: 1 family
- Course
- Progression: Mild
- Mild disease: Survival to adulthood
- Death: 2 patients (2 & 5 months)
- Laboratory
- Muscle pathology
- Internal nuclei
- Type I predominance
- Cores: Multiple; Small; 1 to 2 sarcomeres in length
- Ultrastructure: Abnormal Myofibrillar structure; Z-band streaming
- EMG: Myopathic
- Muscle MRI
- Fatty atrophy
- Locations
- Legs: Anterior & Posterior
- Sartorius > Gracilis
- Quadriceps: Spared
- Serum CK: Normal
- Muscle pathology
Congenital myopathy with excess of thin filaments 5
● α-Actin (ACTA1; Skeletal muscle)- Genetics
- Actin (ACTA1) gene mutations
- Gly15Arg: Surface exposed residue
- Val163Leu: Hydrophobic core
- Allelic syndromes
- Actin (ACTA1) gene mutations
- Clinical
- Hypotonia
- Respiratory insufficiency
- Cardiomyopathy
- Course: Variable with same actin mutation
- Death in 1st months: 2 patients
- Survival to childhood: 1 patient
- Serum CK: Normal
- Muscle
- Masses of thin filaments (Actin)
- Nuclear ± cytoplasmic rods
- Fiber size variation
Carey-Fineman-Ziter Syndrome 1 (CFZS 1)
143
●
Transmembrane protein 8C (TMEM8C; Myomaker; MYMK)
- Nosology: Myopathy, congenital nonprogressive, with Möbius sequence & Robin sequence
- Epidemiology: > 25 patients
- Genetics
- Mutations: Missense
- Also see: CFZ2
- TMEM8C protein
- Myogenesis
- Role in: Fusion of mononuclear myoblasts to form multinucleate myocyte syncytia
- Mutations: Satellite cell disorder
- Other satellite cell disorders: MYOSCO; LGMDR21
- Clinical
- Onset: Congenital
- Myopathy
- Developmental delay
- Hypotonia
- Non-progressive
- Muscle hypoplasia
- Pectoral & Humeral muscles (Poland) (50%)
- Often bilateral
- Weakness
- Axial & Appendicular
- Mild
- Independent mobility in childhood
- Respiratory: Restrictive disease in 4th decade
- Mobius syndrome
- Ocular
- Ophthalmoparesis
- Mild limitations at extremes of gaze
- No VI paresis
- Ptosis
- Palpebral fissures: Down slanted
- Ophthalmoparesis
- VII nerve: Facial weakness, bilateral
- X nerve
- Dysphagia
- Feeding disorders (100%)
- Ocular
- Craniofacial
- Macrocephaly
- Nose: Triangular; Upturned/broad
- Robin sequence
- Micrognathia/Retrognathia
- Glossoptosis: Tongue displaced backward; May obstruct airway
- Cleft palate (50%)
- Skeletal
- Scoliosis (40%)
- Talipes (40%)
- Equinovarus
- Brachydactyly (70%)
- Contractures: Congenital
- CNS
- Usually normal intelligence
- Cryptorchidism in males
- Pulmonary hypertension: Adults
- Laboratory
- Brain MRI: Normal
- Serum CK: Mildly high
- Electrophysiology
- NCV: Normal
- EMG: Myopathic (Small motor units); Fasciculations
- Serum CK: Normal
- Muscle biopsy
- Fiber size variation
- Degeneration
- Regeneration (Desmin + fibers)
- Fiber types
- Type II predominance & Hypertrophy
- Type I: Small
- Myogenin positive nuclei: Few fibers
- Replacement by fat: Older patients
- Muscle MRI
- Most involved: Adductor magnus; Sartorius
- Other involved: Paraspinal; Gluteus maximus
Carey-Fineman-Ziter Syndrome 2 (CFZS2)
●
Myomixer (MYMX)
- Epidemiology: 1 family, 2 siblings
- Genetics
- Mutation: Homozygous; Nonsense; Arg46*
- MYMX protein
179
- Muscle development: Upregulated
- Myogenesis
- Membrane protein
- Synergistic with: Myomaker
- Sonic Hedgehog: Inhibits MYMX expression
- Mutation: Impairs muscle cell fusion
- Clinical
- Onset age: 1st decade
- Face
- Weakness
- Reduced movement
- Dysmorphism: Micrognathia
- Motor
- Axial hypotonia
- Weakness: Moderate; Proximal & Distal
- Exercise intolerance
- Scoliosis: Progressive
- CNS: Normal cognition
- Laboratory
- Muscle biopsy: Minimal patholopgy
- Muscle ultrasound: Normal
Reducing Body Myopathies 3, 4
● Four-and-a-half-LIM protein 1 (FHL1)- Genetics
- Mutations: Missense; H123Y, H123Q, C132F, C153R, C153Y
- General location: Zinc coordinating residues in second LIM domain, or 1st part of LIM3 domain
- Histidine 123 mutations: Often de novo
- Zinc coordinating residue cysteine 153: Milder phenotype
- Allelic disorders
- FHL1 protein
- Cellular location
- Myoblasts: Nuclear
- Differentiated myotubes: Cytosolic
- Mutation effects
- Induces formation of aggresome-like inclusions
- Inclusions incorporate both mutant & wild-type FHL1
- Other proteins trapped in a dominant-negative manner
- Cellular location
- Clinical syndromes
75
- Reducing body myopathy: Severe; Early onset (RBMX1A)
- FHL1 Genetics
- Family history: Often sporadic
- Mutations: Missense; His123Tyr, Cys132Phe, Cys150Tyr
- Clinical
- Onset
- 1st 4 years
- Most severe: Motor delay in 1st year
- Not congenital
- Weakness
- Proximal > Distal
- Progression: Rapid; May occur after stable phase
- Respiratory
- Asymmetric
- Skeletal
- Rigid spine: Some patients
- Scoliosis
- Contractures: Variable
- Later in disease course
- Dysphagia
- Face: Ptosis
- Fatal outcome: < 10 years in some patients
- Onset
- Laboratory
- FHL1 Genetics
- Reducing body myopathy: Late onset; Childhood & Adult (RBMX1B; RBMX2)
6
- FHL1 Genetics
- Inheritance: Dominant
- Mutations: Missense; Cys153Tyr, Cys153Arg
- Clinical
- Onset age
- Up to 4th decade
- Early development: Normal
- Weakness
- Proximal > Distal
- Scapuloperoneal
- Asymmetric
- Respiratory failure
- Paraspinal muscles: May be involved
- Progressive to diffuse involvement
- More prominent than in parents
- Skeletal: Less than with childhood onset
- Rigid spine: Some patients
- Scoliosis
- Cardiomyopathy: Dilated; Some patients
- Carrier females
- May be symptomatic
- More signs with skewed X-inactivation
- Onset age
- Laboratory
- Serum CK: Increased to 3x normal
- EMG
- Mixed neurogenic & myogenic patterns
- May show spontaneous activity
- FHL1 Genetics
- Reducing body myopathy: Females, Young + Rapid progression
212
- Epidemiology: 5 patients
- Genetics
- Inheritance: Often Dominant, de novo
- Mutations: Missense; Cys104Arg, Cys109Tyr, His123Gln, Cys150Arg, Cys153Ser
- Clinical
- Onset age: Mean 13 years; Range 3 to 21 years
- Weakness
- Asymmetric
- Proximal > Distal
- Respiratory
- Course: Rapidly progressive to loss of ambulation (Mean 6 years)
- Laboratory
- Serum CK: Mean 500 to 600
- Muscle pathology
- Reducing bodies: Around nuclei & lipid droplets; Within protein aggregates
- FHL1 & Desmin (Eosinophilic) aggregates
- Autophagic material
- Sarcomere disorganization
- Muscle MRI: Asymmetric, Proximal > Distal; Paravertebral
- Scapuloperoneal (Hyaline body) myopathy, Type I
- Congenital myopathic: Uncommon
- Delayed developmental milestones
- Benign course
- ? Different disorder
- Reducing body myopathy: Severe; Early onset (RBMX1A)
- Muscle biopsy
- Muscle fiber size: Variable
- Increased Acid phosphatase activity
- Reducing bodies
- Location: Cytoplasmic; Often near nucleus
- Histochemistry staining
- Trichrome
- Dark green inclusions
- ± Associated with eosinophilic cytoplasmic bodies
- Menadione-NBT (without α-glycerophosphate): Positive
- Mechanism: Reduce nitro-blue tetrazolium (NBT)
- Pyronin
- Trichrome
- Contain aggresome-like proteins
- Desmin
- Ubiquitin
- Luminal endoplasmic reticulum chaperone GRP78
- γ-tubulin
- FHL1
- MYBPC1
- Pericentrin
- May be more common in type I muscle fibers
- Rimmed vacuoles in some patients
- Ultrastructure
- Reducing bodies
- Structure: Granular tubulo-filamentous material (17 nm in diameter)
- Location: Periphery of sarcoplasm & Around nucleus
- Cytoplasmic bodies: Common
- Reducing bodies
- Immunocytochemistry: Reducing bodies
- Contain proteins normally associated with nucleoli
- Also stain for dystrophin, α-sarcoglycan, vimentin & ubiquitin
- Other disorders with reducing bodies
Fingerprint Body Myopathy
- Epidemiology: Few sporadic cases
- Genetics
- Inheritance: Sporadic or Recessive
- Clinical
- Onset: Congenital
- Weakness
- Proximal > Distal: Limbs & Trunk
- Symmetric
- Sparing of cranial nerves
- Hypotonia
- Intellectual impairment: Occasional
- Course: Nonprogressive or Slowly progressive
- Laboratory
- Serum CK: High in some patients
- EMG: Myopathic
- Muscle biopsy: Inclusions
- Histochemistry: Ovoid; 1 to 10 μM; Red on H& E; Green on GT
- Ultrastructure: Arrays of parallel osmiophilic lamellae resembling fingerprints
- Fingerprint bodies also seen with
Congenital myopathy with apoptotic changes 9
- Epidemiology: 1 Sporadic Japanese patient identified
- Clinical
- Onset: Congenital hypotonia
- Psychomotor development: Markedly delayed
- Motor
- Hypotonia
- Muscle atrophy: Generalized
- Myopathic face
- Tendon reflexes: Normal
- Contractures: Ankles
- Laboratory
- Serum CK: Normal
- Serum Lactate: Normal
- Muscle pathology
- Muscle fibers
- Small
- Basophilic: Scattered
- Acid phosphatase positive: Many
- Immature (Type 2C): 40%
- Vacuoles: 20% of fibers
- Endomysial fibrosis: Mild
- Nuclei: Condensation of chromatin; Fragmentation
- Apoptosis related changes
- Bax-positive (Pro-apoptotic) muscle fibers: 80%
- No Bcl-2 positive (Anti-apoptotic) muscle fibers
- Activated caspase-3 in fibers: 6%
- TUNEL positive nuclei: 2%
- Muscle fibers
Hyaline Body Myopathies
|
MYH7: 14q11.2 MSMA: Dominant MSMB (Scapuloperoneal 3): Recessive Scapuloperoneal 2: Dominant Scapuloperoneal 1: FHL1; Xq26; Dominant |
Hyaline Body Myopathy (CMYO7A)
● Myosin - Cardiac β heavy chain (MYH7)
- Nosology: Also called
- Genetics
- Mutation
- Missense
- Arg1845Trp; His1904Leu
- Location
- Rod region of slow/β-cardiac MyHC
- Exons 37 to 39
- Heterozygous
- Missense
- Allelic disorders: MYH7 mutations cause
- Cardiomyopathy: Mutations commonly in globular head domain
- Hypertrophic cardiomyopathy (CMH1)
- Hypertrophic cardiomyopathy with cores
- Dilated cardiomyopathy (CMD 1S)
- Left ventricular non-compaction 5 (LVNCS5)
- Mutation locations: Globular head, ATP binding pocket, Exons 8�9
- Congenital Heart Disease: Septal defects; Ebstein anomaly 175
- Myopathy: Mutations in rod & tail domains (Exons 32 to 39)
- Distal myopathy (MPD1; Gowers-Laing)
- Scapuloperoneal myopathy: Arg1845Trp
- Multicore/Minicore myopathy: Child onset; Axial
- Hyaline body myopathy (Myosin storage)
- Cytoplasmic bodies
- Congenital fiber type disproportion (CFTD): Dominant
- Neonatal hypotonia
- Myopathy + Cardiomyopathy
- Cardiomyopathy: Mutations commonly in globular head domain
- Mutation
- MYH7 protein
- Clinical features
- Variable penetrance
- Onset: Infancy or early childhood
- Motor
- Early
- Hypotonia
- Delayed motor milestones
- Weakness
- Proximal ± Distal
- Scapuloperoneal
- Arms & Legs
- Symmetric
- May be severe & disabling
- Early
- Course: Slowly or Non-progressive
- Systemic: Occasional cardiac arrhythmias
- Laboratory
- Serum CK: Normal or Mildly elevated
- EMG: Myopathic
- Muscle biopsy
- Hyaline bodies
- Subsarcolemmal
- Eosinophilic
- Mostly in Type I fibers
- Lack oxidative or glycogen activity
- May stain for myosin ATPase (Acid pH 4.3)
- Ultrastructure
- Matrix of granular & filamentous material
- May be subsarcolemmal
- Not membrane bound
- Some intranuclear inclusions
- Type I muscle fiber predominance
- Hyaline bodies
- Hyaline bodies also noted in an adult onset scapuloperoneal myopathy
- MYH7 variant: Multi-minicore Disease (MmD) with Cervical + Axial involvement ± Cardiac
94
- Epidemiology: 10 families
- Genetics
- Inheritance: Dominant; Sporadic; Mosaic
- Mutations: Missense; Proximal rod domain (Exons 31, 32 & 34)
- Clinical
- Onset age: Childhood
- Weakness
- Proximal or Distal predominant
- Arms & Legs
- Respiratory: With disease progression
- Face: Mild
- Scapular winging
- Trunk: Posterior neck & Spine
- Asymmetry: Mild
- Course: Slow progression
- Wasting: Forearms
- Myalgias
- Cardiac
- Cardiomyopathy: Dilated
- Sudden death in 1 family
- Tremor
- Spinal rigidity
- Contractures: Elbows; Long finger flexors
- Laboratory
- Muscle biopsy
- Type 1 fiber predominance
- Internal nuclei
- Core-like structures (> 50%): Multicores; Eccentric cores
- Ring fibers
- Ultrastructure: Myofibrillar changes
- MRI
- Axial: Tongue; Neck & Thoracic extensors; Sternomastoid
- Arm: Posterior relatively spared
- Hip: Gluteus minimus; Tensor fascia lateae
- Leg
- Distal > Proximal
- Posterior > Anterior
- Thigh: Adductor longus; Sartorius; Gracilis; Biceps femoris, short head
- Distal leg: Soleus; Tibialis anterior; Extensor digitorum longus
- "Inverted collagen VI sign"
- Band of fibroadipose infiltration within relatively preserved muscle tissue
- Most prominent: Soleus muscle
- Specificity vs LMNA & SEPN1: Signal in Tibialis anterior, Gluteus minimus, Peronei, Tibialis posterior
- NCV & EMG: Non-diagnostic or Myopathic
- Serum CK: Usually normal; May be mildly high
- Muscle biopsy
- MYH7 variant: Congenital fiber type disproportion
- Epidemiology: 1 family, 7 patients
- Genetics
- Inheritance: Dominant
- Mutation: c.5807A>G
- MYH7 protein
- Clinical
- Onset
- Age: Infancy to Childhood
- Hypotonia or Gait disorder
- Weakness
- Proximal > Distal
- Gait: Waddling
- Foot drop: Some patients
- Progression: Slow; Ambulation maintained
- Muscle atrophy: Proximal
- Tendon reflexes: Reduced except at ankles
- Cardiac: Normal
- CNS: Normal
- Onset
- Laboratory
- Electrodiagnostic
- EMG: Myopathic; Fibrillations & Positive sharp waves
- NCV: Normal
- Muscle MRI
- Abnormal: Paraspinal & Proximal limb; Symmetric
- Spared: Gracilis; Semitendinosus
- Muscle pathology
- Type I muscle fibers: Small; Predominant
- MYH7 aggregates: Subsarcolemmal; In older patient
- Electrodiagnostic
Congenital Myopathy with Muscle Spindle Excess (CMEMS)
18
●
V-HA-RAS Harvey rat sarcoma viral oncogene homolog (HRAS)
- Nosology: Costello syndrome
- Epidemiology: 5 patients with selective myopathy
- Genetics
- Mutations: Missense; Gln 22 Lys, Glu63Lys
- Allelic with
- Costello syndrome: Multiple congenital anomalies
- Faciocutaneoskeletal syndrome (FCSS)
- Schimmelpenning-Feuerstein-Mims syndrome, somatic mosaic
- Somatic mutations in neoplasms
- HRAS protein
- Location: Cell membrane & Golgi
- Binds GDP/GTP
- Has intrinsic GTPase activity
- Activated by a guanine nucleotide-exchange factor (GEF)
- Inactivated by a GTPase-activating protein (GAP)
- Interacts with: EGR3
- Clinical
- Onset
- Age: Birth or Prenatal
- Hypotonia
- Arthrogryposis
- Prenatal
- Reduced movements
- Polyhydramnios
- Weakness
- Diffuse
- Respiratory failure
- Areflexia
- Face: Coarse; Frontal hollowing; Flaccid cheeks; Triangular mouth; High arched palate
- Skeletal & Joints
- Arthrogryposis
- Distal laxity
- Short stature
- Cardiomyopathy
- Atrial tachycardia
- Hypertrophic cardiomyopathy
- Heart failure
- May produce early death
- Organomegaly
- Neoplasms
- Neuroblastoma: Congenital; Adrenal
- Papillomata: Skin; Hands & Feet
- Skin: Loose & redundant over hands & feet
- Course (Variable): Death at 14 months to Survival at 5 years
- Onset
- Laboratory
- Metabolic studies: Normal
- Brain MRI: Enlarged subarachnoid space & ventricles
- EMG, RNS & NCV: Normal or Fibrillations & Small MUPs
- Muscle pathology
- Myopathy
- Muscle fibers (Variable)
- Scattered atrophy to Severe loss
- Type I small
- Muscle fibers (Variable)
- Muscle spindles: Excessive number
- Number: 1 or more per fascicle
- Muscle: Especially in deltoid & quadriceps
- Intrafusil muscle fibers
- Abnormal shapes
- Size: Hypertrophy
- Nuclei: Dysmorphic
- Number per spindle: 0 to 17
- Type: I or II
- May replace most extrafusil muscle fibers
- Muscle spindles: Other disorders
Congenital Myopathy with Caps
57
|
Genetics: General Mutations in some of same genes that produce Rod myopathies & CFTD Dominant 1: γ-Tropomyosin 3 (TPM3): 1q22 2: β-Tropomyosin 2 (TPM2): 9p13 α-Actin 1, Skeletal muscle (ACTA1): 1q42 Recessive Nebulin: 2q23 Myopalladin: 10q21 TRIP4: 15q22 Muscle Pathology Also see Myofibrillar (Desmin storage) myopathy Cap pathology |
Cap Myopathy 2 (CAPM2)
● β-Tropomyosin 2 (TPM2)
- Epidemiology: ~8 sporadic cases reported
- Genetics: TPM2
- Mutations: Inframe deletion or Missense (K7del, Glu139del, Asn202Lys); Heterozygous
- TPM2 allelic disorders
- Mutations not present in: Recessive cap myopathy patients
- TPM2 protein
- Clinical
- Onset: Congenital or Childhood
- Motor
- Hypotonia
- Weakness
- Proximal & Distal
- Face
- Respiratory
- Respiratory insufficiency: Severe cases
- Course
- Slowly progressive weakness
- Severe disease: Death at 14 years in 1 patient
- Skeletal
- Contractures: Especially distal
- Face: Long narrow
- Palate: High arched
- Chest: Deformity; Scoliosis
- Severity: Variable
- Laboratory
- Serum CK: Mildly elevated or Normal
- EMG: Myopathic
- Muscle pathology
- Caps
- Staining: Dark on Gomori trichrome
- More common in type I muscle fibers
- Regions at periphery of muscle fibers
- Stain for: NADH; Esterase; Desmin; α-actinin; tropomyosin; Actin; SERCA2; Nebulin; Myotilin
- Ultrastructure
- Abnormal Z-line material
- Disorganized myofibrillar architecture
- Nemaline rods
- Type I muscle fibers: Small
- Internal nuclei
- Caps
Myofibrillar myopathy 11 (MFM11): Myopathy with Eccentric cores
156
●
UNC45 myosin chaperone B (UNC45B)
- Epidemiology: 9 families; 11 patients
- Genetics
- Mutations
- Common: Arg754Gln
- Other: Missense; Splice
- No biallelic null
- Allelic disorder: Cataract 43
- UNC45A
: Osteootohepatoenteric syndrome
- Mutations
- UNC45 protein
- Myosin-specific chaperone
- Expression: Skeletal muscle
- Myosin assembly: with HSP90
- Required for myosin thick filament assembly & stability in cardiac & skeletal muscle
- Myoblast fusion
- Sarcomere organization
- May contribute to myosin degradation via ubiquitin/proteasome system
- Loss-of-function mutations
- Paralysis & Cardiac dysfunction
- Reduced muscle myosin accumulation
- Defective thick filament assembly
- Clinical
- Onset: Congenital to 6 years
- Hypotonia
- Weakness: Proximal; Axial; Gowers sign; Respiratory
- Muscle size: Calf hypertrophy
- Voice: Nasal
- Contractures: Ankle
- Normal: Face; EOM
- Course: Slow decline or improvement
- Heart: Branch block or Normal
- Laboratory
- Muscle biopsy
- Muscle fibers
- Core-like lesions
- Complex
- Pale
- Location: Eccentric; Central or Subsarcolemmal regions
- Shape: Irregular
- Size: Large
- Frequency: Common
- ATPase activity: Altered
- Ultrastructure: Z-band streaming; No mitochondria; Unstructured
- Some neonatal myosin +
- Type 1 predominance
- Subsarcolemmal organelles
- Cytoplasmic bodies
- Core-like lesions
- Muscle fibers
- Serum CK: Normal
- EMG: Myopathic
- Muscle MRI
- Semimembranosus relatively spared
- Fat infiltration in all thigh muscles
- Muscle biopsy
Myofibrillar Myopathy 8 (MFM8): Early-Onset with Internal Nuclei, & Myofibrillar Disorganization
133
●
Pyridine nucleotide-disulfide oxidoreductase domain-containing protein 1 (PYROXD1; PYRD1)
- Epidemiology: 17 families, 24 patients
- Genetics
- Mutations
- Missense: Common; Asn155Ser recurrent
- Some splice site & stop: More severe & Earlier disease onset
- None with 2 loss of function mutations
- Allelic disorder: LGMD syndrome, Adult onset
- Mutations
- PYROXD1 protein
- Oxidoreductase: Class 1
- Location: Nuclear; Cytoplasm (Striated; Related to Desmin)
- Functions
- Mitochondrial respiratory chain
- Regulation of tRNA ligase complex
- Clinical
- Onset age: Congenital to 8 years
- Weakness
- Distribution
- Symmetric
- Arms; Legs; Axial & Neck; Face
- Proximal > Distal: Some patients
- Bulbar: Dysphagia 50%; Nasal speech
- Ptosis, mild 33%
- Scapular winging: Mild 50%
- Respiratory
- Severity: Moderate
- Course: Slow progression
- Distribution
- Eyes: Blue sclerae
- Tendon reflexes: Reduced
- Joints
- Contractures 20%
- Hypermobility: Distal + other
- Skeletal: Some with
- Scoliosis
- Spinal rigidity: Thoracolumbar
- Pes planus
- Osteopenia
- Skin: Soft
- Recurent infections 50%
- Cardiac disorders; Oldest patient
- Laboratory
- Serum CK: Normal to 1050
- Urine: Deoxypyridinoline (DPD) high
- Muscle pathology
- Fiber size: Varied
- Small muscle fibers: Polygonal; Some with large internal nusclei
- Internal architecture: Cores; Irregular
- Mitochondrial distribution: Abnormal
- Myofibrillar inclusions: Desmin; Myotilin; α-Actin; αB-Crystallin
- Sarcomere disorganization: Z-band streaming
- Thin filament accumulations
- Rods 25%
- Nuclei: Internal; Often in clusters; Large
- Endomysial connective tissue: Mildly increased
- PYROXD1 protein: Normal or reduced levels, Not absent
- HSP70 & Glutathione reductase: Increased on Western blot
- NCV: Axonal sensory neuropathy in 1 family
- Repetitive nerve stimulation: Decrement
- MRI: Generalized, Symmetric; Proximal legs
- PYROXD1 variant: LGMD syndrome, Adult onset
147
- Epidemiology: 4 Finnish patients
- Genetics
- Inheritance: Recessive
- Mutations: Asn155Ser homozygous or with Tyr354Ser
- Clinical
- Onset age: 3rd to 5th decade
- Weakness: Proximal; Legs > Arms; Respiratory
- Muscle wasting
- Cardiac: Normal
- Peripheral nerve: Normal
- Course: Slow progression; Ambulant past 60 years in many
- Laboratory
- Muscle MRI: Anterolateral thigh muscles involved
- Serum CK: Normal to 350
- Muscle: Dystrophic
- Fiber size: Varied; Hypertrophy & Atrophy
- Internal nuclei
- Endomysial connective tissue: Increased
- No myofibrillar pathology
X-linked Mental retardation syndrome (Cabezas)
14
●
Cullin 4B (CUL4B)
- Genetics: Missense & Stop mutations
- CUL 4B protein
- Ubiquitin ligase
- Complexes with dioxin receptor (AHR)
- Onset
- Birth to Childhood
- CNS: Low weight; Delayed milestones
- Skeletal: Short stature; Small feet; Gap between 1st & 2nd toes; Hyperextensible joints; Kyphosis
- Obesity
- Small testes
- Prominent lower lip
- Neurological
- Tremor: Fine; Upper limbs
- Fine motor coodination: Reduced
- Mental retardation
- Behavioral: Reduced Attention span; Hyperactivity; Mood swings; Aggression
- Motor: Wide based gait; Wheelchair needed
- Muscle wasting: Lower legs
Muscular Dystrophy, Congenital, with Rapid Progression (MDRP)
●
BET1 Golgi vesicular membrane-trafficking protein (BET1)
- Epidemiology: 3 patients; 2 families
- Genetics
- Mutations: Missense; Stop
- BET1 protein
- Clinical
- Onset: Prenatal or Neonatal
- Weakness: Diffuse; Respiratory
- Extraocular movements: Restricted
- Skeletal: Scoliosis; Joint contractures
- CNS: Speech delay; Seizures
- Course: Progressive; Death in infancy or childhood
- Laboratory
- Serum CK: High
- Muscle
- Fiber sizes: Varied
- Endomysial connective tissue: Increased
- Type 1 fibers: Small
- α-dystroglycan: Reduced glycosylation
- Brain MRI: Normal or Corpus callosum Δ
Congenital Weakness with Diarrhea & Deafness
●
Recessive
- Clinical
- Weakness
- Hypotonia at birth: Feeding difficulties
- Myopathic faces
- Tendon reflexes: Reduced
- Improvement with age
- Joint contractures
- Diarrhea: Episodic with intercurrent infections; Secretory
- Skin: Bullous eruptions; Resolves by 2 months
- Deafness: Sensorineural
- Microcephaly
- Cryptorchidism: In male
- Weakness
- Laboratory
- Muscle pathology: Scattered, small muscle fibes, often type II
- Diarrhea-associated: Zinc deficiency; Electrolyte disorders
- Alkaline phosphatase: Low
Congenital Fiber Type Size Disproportion (Type I fibers small)
51
|
Differential diagnosis General CFTD 1: α-Actin: 1q42; Dominant CFTD 2: Xq13; Recessive CFTD 3: SEPN1: 1p36; Recessive CFTD 4: TPM3; 1q21; Dominant CFTD 5: TPM2; 9p13; Dominant CFTD: MYL2; 12q24; Recessive CFTD: HACD1 (PTPLA); 10p12; Recessive CFTD: MYH7; 14q11; Dominant CFTD: MYL2; 12q24; Recessive CFTD: Lamin A/C Myopathy SCN4A: 17q23; Recessive Titin: 2q31; Recessive Also see: Fiber type predominance RYR1 (Type 1) Pathology |
- Definition
- Onset
- Age
- Congenital or Childhood
- Asymptomatic (4%)
- Weakness
- Age
- Muscle biopsy pathology: Bimodal distribution of fiber sizes
- Criterion: Type 1 muscle fibers 12% smaller diameter than Type 2
- Onset
- Clinical: General features
- Family history: 43%; Dominant or Recessive
- Motor
- Weakness
- Extremities (95%): Diffuse or Proximal > Distal
- Degree: Mild to Severe (27%)
- Face (40%)
- Bulbar (35%): Associated with more severe weakness
- Respiratory (28%)
- Hypotonia
- Weakness
- Tendon reflexes: Reduced or Absent
- Ocular
- Ophthalmoplegia (19%): Worse prognosis
- Intelligence: Normal
- Skeletal (25%)
- Arthrogryposis
- Scoliosis
- Course: Static (55%); Improving (36%); Slow progression (9%)
- Laboratory
- Serum CK: Normal or < 3x upperlimit of normal
- EMG: myopathic or some Neuropathic features
- Muscle pathology
- Fiber size variation
- Degree: Mean = Type 1 fibers 41% smaller diameter than Type 2; Range up to 74%
- Correlation: Larger size disparity with more severe clinical features
- Type 2B fibers < 5% or absent: 35%
- Type 1 fiber predominance (50%)
- Internal or central nuclei (> 5%): 22%
- Nemaline rods (6%)
- Fiber size variation
- CFTD 1
178
● α-Actin (ACTA1);
Chromosome 1q42.13; Dominant or de novo
- Epidemiology: 9 patients
- Genetics
- Mutations
- Heterozygous
- Missense: Leu221Pro; Asp292Val; Met327Lys; Pro332Ser
- Different from rod myopathy mutations
- Location: Actin surface swept by tropomyosin during muscle activity
- Frequency in CFTD: Unusual; 6%
- Other α-Actin disorders
- Mutations
- Clinical: Severe phenotype
- Onset: Congenital; Weakness
- Weakness
- Distribution: Diffuse
- Respiratory
- Severe
- Cardiomyopathy, Dilated: 20%
- No ophthalmoplegia
- Course: Respiratory failure; Death < 4 years (2 of 3 patients
- Muscle pathology
- Type 1 muscle fibers: Small; 45% to 54% of Type 2
- No rods
● Chromosome Xq13.1-q22.1; Recessive
- Epidemiology: Australian family
- Onset
- Age: Congenital
- Hypotonia & Weakness
- Clinical
- Weakness
- Ptosis: Bilateral
- Face
- Suck & cry: Weak cry
- Hypotonia: Generalized
- Respiratory insufficiency
- Cardiomyopathy: Dilated; In one survivor
- Course
- Death in most: Respiratory failure; Age 6 to 14 weeks
- Survivor: Facial & mild generalized weakness
- Weakness
- Laboratory
- Serum CK: Normal
- Lactate: Normal
- Muscle biopsy: Type 1 muscle fiber smallness
- Carriers
- Mild weakness: General & Face (Frontalis)
● Selenoprotein-N, 1 (SEPN1; SELENON)
- Genetics
- Common mutation: G315S
- Allelic disorders
- Clinical
- SEPN1-like syndrome
- Truncal hypotonia: Early
- Strength
- Neck weakness: Early
- Respiratory impairment: Adolescence to early adulthood
- Limb strength: Relatively preserved
- Scoliosis
- Progressive
- Mid childhood to Adolescence
- Relative lordosis of upper spine
- Insulin resistance
- SEPN1-like syndrome
- Muscle histology
- Mean type 1 fiber diameter more than 12% smaller than type 2 fiber diameter
- No multiminicore lesions
● Myosin, light chain 2, regulatory, Cardiac, Slow (MYL2)
- Epidemiology: Dutch & Italian patients
- Genetics
- Mutations: Located in C-terminal exon; Deletion, Splice & Stop
- Allelic disorders
- Hypertrophic cardiomyopathy 10 (CMH10)
- Cardiomyopathy: Familial dilated with Type 1 muscle fiber hypotrophy
- MYL2 protein
- Spindle: Intrafusil muscle fibers
- Myosin regulatory light chain (MLC-2V)
- Bind, with essential light chains, to flexible neck region of the myosin heavy chain
- Structural & regulatory role in muscle contraction
- Clinical
- Onset age: Infancy
- Hypotonia & Weakness: Generalized
- Face
- Mouth: Tented
- Ptosis: Some patients
- Tremor/Clonus: Generalized
- Cardiomyopathy: Dilated
- Death: 4 to 6 months
- Laboratory
- Muscle
- Type 1 muscle fibers: Small (48% to 79% of diameter of type 2)
- NADH: 3 fiber types; Smallest fibers dark
- 2C fibers: Mildly increased
- MYL2: Mild expression in all muscle fibers; Normally absent
- Disorganized sarcomeres
- Myofibrils: Partial loss
- Serum CK: Often normal; May be increased
- Muscle
● Sodium Channel - α subunit (SCN4A; Nav1.4)
- Nosology: CMYP22; CMYO22
- Epidemiology: 6 families; 11 patients
- Genetics
- Mutations
- Allelic disorders
- SCN4A protein
- Clinical syndromes
- Fetal hypokinesia (CMYP22B
)
- Polyhydramnios
- Respiratory failure
- Face: Dysmorphic
- Joint contractures
- Death: 3rd trimester (< 36 weeks; Most) or Shortly after birth
- Congenital myopathy (CMYP22A
)
- Onset age: Congenital or Neonatal
- Hypotonia
- Weakness
- Face
- Neck: Flexion
- Respiratory
- Swallowing
- Axial
- Limbs: Arms & Legs; Proximal > Distal
- Symmetric
- Episodic: Some patients
- Fatigue: Weakness worse with exercise
- Weakness after fever
- Muscle atrophy
- Skeletal
- Spine deformities: Scoliosis
- Face: Elongated
- Palate: High arched
- Scapular winging
- Contractures: Legs
- Craniostenosis: 2 patients
- Tendon reflexes: Normal
- Course
- Improvement in 1st decade
- Subsequent decline
- Fetal hypokinesia (CMYP22B
- Laboratory
- MRI changes: Symmetric; Gluteus maximus, Sartorius, Adductor magnus, Soleus
- EMG: Myopathic; No Myotonia
- RNS: 3 Hz no decrement; 10 Hz decrement in 1 patient
- Muscle
- Fiber size: Varied
- Fat replacement
- Congenital myopathy: Type 1 small & predominant
- Fetal hypokinesia: Type 2 predominant; Hypoplasia
- Corona muscle fibers (1 family)
- Central pallor: NADH
- Whorled
- Nuclei: Internal; Around internal pale regions
- Mitochondria: Clustered at fiber periphery
- Ultrastructure: Myofibrillar disarray; Paracrystalline "Parking lot" inclusions
- Brain MRI: Normal
- Serum CK: Normal to 600
Multisystem Selenoprotein Deficiency 83
● Selenocysteine insertion sequence�binding protein 2 (SECISBP2; SBP2)- Epidemiology: 2 families
- Genetics
- Premature termination, Splicing & Missense mutations
- Mutations: fs255X; C691R
- Allelic disorder: Thyroid hormone metabolism, abnormal (THMA)
- SECISBP2 protein
- Function: Translational incorporation of Selenocysteine during selenoprotein synthesis
- Interaction of a stemloop RNA structure with a multiprotein complex, which includes SECISBP2
- Leads to Selenocysteine incorporation mediated by selenocysteyl-transfer RNA (tRNA[Ser]Sec) at UGA codons
- Disease
- Markedly reduced, but not absent, production of seleoproteins
- Selenium
- Function: Translational incorporation of Selenocysteine during selenoprotein synthesis
- Clinical
- Onset
- Age: Infancy & Childhood
- Developmental delay
- Muscle
- Weakness
- Axial
- Proximal limbs
- Respiratory: Vital capacity reduced
- Focal: Thigh adductors; Sartorius
- Weakness
- Fatigue
- Onset
- Azoospermia: Spermatogenic maturation arrest
- Skin
- Raynaud's
- Cutaneous photosensitivity: Enhanced UV-mediated oxidative damage
- Hearing loss: Sensorineural
- Spine: Stiff; Straight
- Serum CK: Mildly high to 900
- Thyroid: T4 high; T3 normal; TSH normal
- Plasma selenium: Low
- Hematology
- RBC & Total lymphocyte counts: Mild reduction
- Defective T-cell proliferation
- Anti-oxidant mechanisms
- Reduced
- Reactive oxygen species, cellular: Increased
- Telomeres: Reduced length
- Membrane lipid peroxidation: Increased
- Oxidative DNA damage
- Skin: Deficiency of GPx1, TrxR, and MSRB
- Selenoprotein levels: Reduced; Including SEPN1
- Fat mass: Increased
- Carbohydrate metabolism
- Insulin sensitivity: Increased
- Fasting hypoglycemia
- Muscle MRI
- Internal nuclei
- Minicores
- Type 1 muscle fiber predominance
- Varied fiber size: Mild
Ehlers-Danlos, Variant with progressive Kyphoscoliosis, Myopathy & Hearing loss (EDSKMH)
90
●
Peptidyl-prolyl cis-trans isomerase 14 (FK506-binding protein 14; FKBP14)
- Epidemiology: 5 European families
- Genetics
- Mutations
- Homozygous or Compound heterozygous
- Frameshift: Insertion; Deletion
- Common: 1 base pair cytosine in 5C-nucleotide stretch in exon 3
- Other Kyphoscoliotic EDS
- EDSKSCL1
: PLOD1
- EDSKSCL1
- Mutations
- FKBP4 protein
- Localization: Endoplasmic reticulum (ER)
- FK506-binding family of peptidyl-prolyl cis-trans isomerases (PPIases)
- Clinical
- Onset: Birth; Hypotonia
- Skeletal
- Kyphoscoliosis: Progressive; May require surgery
- Joints: Hypermobility, Large & Small joints
- Flat feet
- Skin: Hyperelastic; Soft; Follicular hyperkeratosis
- Hearing loss: Sensorineural
- Myopathy
- Infancy: Hypotonia; Poor head control
- Weakness: Improves through infancy
- Motor development: Delayed; Walking at 2 to 4 years
- Muscle atrophy: Hands
- Weakness: Moderate; May progress in 5th decade; No respiratory failure
- Myopia
- Some patients: Cleft palate; Bladder diverticulum; Tricuspid insufficiency
- Differential diagnosis
- Ehlers-Danlos (VIA; VIB): Kyphoscoliotic form
- Ullrich CMD
- Ehlers-Danlos syndromes: Other neuromuscular
- Laboratory
- Pyridinoline excretion: Normal in urine
- Serum CK: Normal or Slightly high (60 to 300)
- EMG: Normal in early childhood; Myopathic later
- Muscle MRI: Abnormal
- Marked: Vastus lateralis, Rectus femorus, soleus
- Mild: Hip flexors, Neck extensors, Shoulder girdle, Paraspinal
- Normal in 1 patient
- Pathology
- Deficient protein in fibroblasts
- Enlarged ER cisterns
- Altered extracellular matrix
- Muscle fibers
- Atrophy
- Varied size
- NADH: Central pallor
- Ultrastructure: Myofibrillar rearrangements; Z-band streaming
- Deficient protein in fibroblasts
Ehlers Danlos, Hypermobile Syndromes
185
General features- Joint hypermobility: 5th finger extendable to > 90°
- Skin hyperextensibility
- Connective tissue fragility
- Genetic associations: Unknown
- Joint: Pain, Instability, Sub-luxations
- Fatigue
- Disorders of proprioception
- Bruising
- Small fiber 184
- Frequency: 61%
- Pattern: Non-length dependent
- Autonomic
- POTS
- Dry eyes, moth & skin
- Cold hands & feet
- Ulnar neuropathy
- Location: Elbow
- Stretch lesion from repeated dislocation
Shwachman�Diamond Syndrome, Neonatal with Myopathy
127
●
SBDS gene
- Nosology: Pancreatic insufficiency & Bone marrow dysfunction
- Epidemiology: 1 Scandinavian child
- Genetics
- Mutations: Intronic (c.258+2T>C) & Missense (c.41A>G, p.Asn14Ser)
- SBDS protein
- Catalyst in translational activation at ribosomal level
- Clinical
- Onset age: Congenital
- Hypotonia
- Respiratory distress
- Skeletal: Thoracic dysplasia
- Hydronephrosis: Bilateral, Mild
- Laboratory
- Hematology: Intermittent leukopenia & thrombocytopenia
- Serum pancreatic amylase: Low
- Fecal elastase: Low
- Liver function tests: Abnormal
- Muscle
- Fiber size: Small
- Central nuclei: Some fibers
- Fiber types
- 2 predominance
- Frequent embryonic & fetal MyHC in fibers
- Intermyofibrillar network: Reduced in some fibers
- Ultrastructure
- Fibers with disrupted sarcomeric structure & thick Z-discs
- Rod-like structures: Occasional
CNM6: Congenital Myopathy with Type I fiber predominance
134
●
Leucine zipper- & Sterile alpha motif-containing kinase (ZAK; MAP3K20)
- Nosology: Centronuclear myopathy 6 (CNM6)
- Epidemiology: 4 families, 13 patients
- Genetics
- Mutations
- Frameshift & Nonsense
- c.490_491delAT; c.515G>A; c.280_281insT; c.456delT
- Allelic disorder: Split-foot malformation with mesoaxial polydactyly (Phe368Cys missense & Inframe deletion)
- Mutations
- ZAK protein
- Mitogen-activated protein kinase kinase kinase 20 (MAP3K20)
- Mitogen activated protein triple kinase
- Serine-threonine kinase
- Actin fibre modulation
- Gene encodes 2 kinases: ZAKα & ZAKβ
- Signal transduction molecule
- MAPKKK (MAP3K) family
- Stress-activated protein kinase signaling cascade
- Proximal sensor of ribosome collisions during ribotoxic stress response (RSR)
- Directly binds to ribosomes
- ZAKβ
- Only isoform expressed in skeletal muscle
- ZAK isoform 2 (MRK-β): Preferentially activates p38γ/ERK6 pathway via MKK3/MKK6
- Senses mechanical stress
- Activated by muscle contraction & cellular compression
- Abundant phosphorylated protein in muscle
- Muscle disease: Homozygous loss of ZAKα & ZAKβ
- Mitogen-activated protein kinase kinase kinase 20 (MAP3K20)
- Clinical
- Onset age: Congenital to 4 years
- Weakness
- Legs ± Arms
- Proximal & Distal
- Respiratory: Vital capacity 63% to 100%
- Face: Normal
- Course: Slow progression
- Muscle, other
- Size: Thigh atrophy; Calf hypertrophy (50%)
- Cramps
- Fatigue
- Skeletal
- Scoliosis (100%)
- Joint laxity (60%)
- Contractures (16%)
- Scapular winging
- Laboratory
- Serum CK: Normal to 1500
- EMG: Myopathic
- EKG: Normal
- Muscle pathology
- Fiber size: Varied; Small fibers round
- Fiber types
- Type 1 predominance
- Type 2: Small
- Nuclei: Internal; May be clustered
- Internal architecture: Irregular; Subsarcolemmal mitochondria
- Rimmed vacuoles (50%)
- Endomysial connective tissue: Increased or Normal
- Ring fibers
- Protein accumulations: Myofibrillar; Myotilin, FLNC, BAG3, Myofibrillar myopathy markers
CMYO14: Congenital Myopathy with Smallness or Absence of Type 2 Muscle Fibers
146
●
Myosin, light polypeptide 1, alkali, skeletal, fast (MYL1)
- Nosology; CMYP14; CMYO14; MYOFTA
- Epidemiology: 5 patients
- Genetics
- Mutations: Homozygous; Missense (c.488T>G; p.Met163Arg), Splice (c.479-2A>G), c.543del, c.334C>T, c.478+1G>A; Intron
- MYL1 Protein
- Myosin light chain: Fast skeletal muscle
- 2 splice isoforms: MLC1F; MLC3F
- MYL disorders
- Clinical
- Polyhydramnios
- Congenital hypotonia
- Weakness
- Severe
- Distribution
- Cranial: Face; Bulbar; Dysphagia
- Respiratory
- Limbs: Arms > Legs; Proximal
- Tendon reflexes: Absent
- Bone fractures
- Course: Death at 7 months
- Laboratory
- EMG: Myopathic
- RNS: Decrement at 3 Hz stimulation
- Serum CK: Normal
- Brain MRI: Normal
- Muscle
- Muscle fibers
- Size: Varied, Hypertrophy & Atrophy
- Type 1 fibers: Predominance; Hypertrophy
- Type 2 fibers: Small or Absent; No 2B fibers
- "Florets" pattern: Small type 2 fibers encircle larger type 1 fibers
- Large fibers: Express slow myosin
- Small fibers: Express Fast & Fetal myosin; Cardiac α-actin, Desmin, NCAM
- MYL1 protein: Absent
- Perimysial & Endomysial fibrosis
- Fatty replacement
- Muscle fibers
Congenital Hypotonia + Muscle Pathology 189
● Mitogen-activated protein kinase 8-interacting protein 3 (MAPK8IP3)- Epidemiology: 1 patient
- Genetics
- Mutations: Compound heterozygous; Splice donor variant (c.1667+2 T>C, Missense (Asp1237Asn)
- Allelic disorder: Neurodevelopmental disorder ± Brain abnormalities (NEDBA), Dominant de novo
- Protein
- MAPK8IP3 gene encodes: JNK-interacting protein 3 (JIP3)
- Removes lysosomes from axons via dynein-mediated retrograde transport
- Regulates position & shape of myocyte nuclei
- Interacts with: JNK signaling system; Kinesin/Dynein proteins
- Clinical
- Onset age: Congenital
- Hypotonia
- Respiratory insufficiency
- Dysphagia
- Skeletal: Joint Contractures: Micrognathia
- Death: 5 months
- Laboratory
- Muscle: Fiber sizes varied; Endomysial connective tissue increased
- EMG: Spontaneous activity; Early recruitment
- Brain MRI: Mild, non-specific changes
- MAPK8IP3 variant: Neurodevelopmental disorder ± Brain abnormalities (NEDBA)
- Epidemiology: 18 patients
- Genetics
- Inheritance: Dominant de novo
- Mutations: Missense
- Clinical
- Intellect: Disability; Autism
- Muscle tone: Decreased or Increased
- Ataxia
- Spasticity: Diplegia
- Laboratory
- Brain imaging: Cerebral atrophy; Cerebellar & Corpus callosum hypoplasia
- Nerve pathology: Axon loss
- NCV: Axon loss
CMYO25: Congenital Myopathy
192
●
Junctophilin 1 (JPH1)
- Epidemiology: 4 patients
- Genetics
- Mutations: Homozygous; Loss of function
- Possible allelic disorder: CMT2 Dominant
- JPH1 protein
- Triad structure formation & maintenance
- Formation of skeletal muscle triad junctions
- Connects sarcoplasmic reticulum & T-tubules
- Interacts with RYR1: Aids release of Ca++
- Other junctophilins
- Clinical
- Onset: Neonatal
- Early: Hypotonia
- Weakness: Generalized
- Eye: Ptosis; Ophthalmoplegia
- Face: Myopathic
- Bulbar: Dysarthria; Dysphagia
- Limbs: Legs > Arms
- Respiratory
- Course: Slow progression
- Other muscle
- Muscle size: Small
- Myalgia
- Fatiguability
- Skeletal: Lordosis; Scoliosis; Ankle contractures
- Tendon reflexes: Absent or Reduced
- No cardiac or CNS
- Laboratory
- Muscle pathology
- Histochemistry: Type 1 fiber predominance
- Ultrastructure: Triads reduced; Sarcoplasmic reticulum structure abnormal
- Muscle pathology
Return to Myopathy & NMJ Index
Return to Neuromuscular Home Page
References
1. Pediatric Neurology 1996;15:150-152
2. Muscle & Nerve 1998;21:40-47, Am J Human Genet 2011; Online June, Neuromuscul Disord 2020;30:47-53, Skelet Muscle 2022;12:23, Neuromuscul Disord 2023;33:589-595
3. J Neurol Sci 1995:128:58-65
4. Neuromuscular Disorders 1998;8:162-168
5. Neuromuscular Disorders 1997;7:160-168
6. Neuromuscular Disorders 1999;9:580-586
7. Am J Hum Genet 2000 February 66:
8. Ann Neurol 2000;47:152-161
9. Ann Neurol 2000;47:531-536
10. Human Mutation 2000;15:393-409, Hum Mutat 2002;19:114-121, Muscle Nerve 2017 Nov 17
11. Human Mutation 2000;15:410-417
12. Neuromuscular Disorders 2000;10:264-273, 2004;14:754�766
13. Am J Hum Genet 2000;67:000, J Med Genet 2020 Sep 29
14. J Med Genet 2000;37:663-668
15. Neuromuscular Disorders 2000;10:541-547
16. Neuromuscular Disorders 2000;10:548-552; 2003;13:January online
17. Hum Mol Genet 2000;9:3083-3090
18. Muscle Nerve 2001;24:138-143, Neuromuscular Disorders 2014; Online June
19. Neurology 2001;56:1059-1069
20. Neuropediatrics 2001;32:107-109
21. PNAS 2001;98:7516-7521, Ann Neurol 2001;50, Am J Hum Genet 2002;70:1446-1458
22. Ann Neurol 2001;50:, Trends Mol Med 2001;7:362-368, J Muscle Res Cell Motil 2019;40:111-126, Neurology 2021 Jan 4, Neuromuscul Disord 2021 Jul 24
23. J Neuropath Exp Neurol 1982;41;298-314
24. Neuropath Appl Neurobiol 1994;20;232-237
25. Neuromuscular Disorders 2001;11:538-541
26. Neuromuscular Disorders 2001;11:635
27. Neuromuscular Disorders 2002;12:13-18, Brain 2003;126 Online June, Am J Human Genet 2010; Online November
28. Br J Plast Surg 2001;54:132-136
29. Developmental Cell 2001;1:717�724
30. Ann Neurol 2002;51:750-759
31. Neuromuscular Disorders 2002;12:392�398
32. J Neuropath Exp Neurol 2002;61:520-530
33. Ann Neurol 2000;48:745-757
34. Neuromusc Disord 2002;12:623-630
35. Neurology 2002;59:613-617
36. J Neurol Sci 2002; Online October, J Neuropathol Exp Neurol 2007;66:57-65, J Neuropathol Exp Neurol 2020;7:1370-1375
37. PNAS 2002;99:15060-15065
38. Neuromuscular Disorders 2002;12:November online
39. Neurology 2003;60:1363�1365
40. Human Molecular Genetics 2003;12:1045�1053
41. Am J Hum Genet 2003; Online July, Human Molecular Genetics 2005;14:279�293
42. Acta Neuropathol 2003;106:137�142
43. Human Molecular Genetics 2003; Online September
44. Ann Neurol 2003;54:494�500
45. Brain 2003;126:2341-2349, Acta Neuropathol Commun 2014;2:148, Am J Med Genet A 2019;179:386-396
46. Nature Genet 2003;Online November
47. Nature Medicine 2004;Online June
48. Curr Opin Neurol 2004;17:205�209
49. Neurology 2004;62:1484�1490
50. Hum Genet 2003;113:297�306, PLoS One 2012;7:e46408
51. J Neuropath Exp Neurol 2003;62:977�989, Ann Neurol 2004; Online October
52. Neuromuscular Disorders 2004;14:785�790
53. Neuromuscular Disorders 2004;14:779�784
54. Human Molecular Genetics 2005;14:295�305
55. Human Genetics 2005; Online May, Neuromuscular Disorders 2012; Online August
56. Neurology 2005;64:1638�1640, Neurology 2019 Sep 20
57. J Neurol Sci 2002;201:27�31, Neuromuscular Disorders 2007; Online April
58. J Med Genet 2005;42;408-415
59. Neurology 2005;65: Online September 7, Neurology 2014 Nov 5, J Neurol 2018;265:542-551, Neurology 2019;93:e298-e305, Neurol Neuroimmunol Neuroinflamm 2022;9:e1184, Curr Neurol Neurosci Rep 2023 Oct 19
60. Nature Genetics 2005; Online October 16, Neurol Genet 2022;8:e200027
61. Ann Neurol 2005; Online Dec
62. Neuromuscular Disorders 2006; Online Jan, Acta Neuropathol Commun 2022;10:101
63. Brain 2006; On-line June
64. Neuromuscular Disorders 2006;16:S62
65. Am J Hum Genet 2006; Online October
66. Ann Neurol 2006; Online December 22
67. Neuromuscul Disord 2007 May 28
68. Nature Genetics 2007; Online August
69. Neurology 2008;70:114�122, Pediatr Neurol 2014; Online Apr
70. Ann Neurol 2008; Online Feb
71. Ann Neurol 2007;62:666-670, Neuromuscular Disorders 2013; Online Feb
72. Science 2008 Jul 24
73. Human Mut 2008; Online Dec
74. Neuromuscular Disorders 2009; Online January
75. Brain 2009 Jan 29
76. Prenat Diagn 2009 Mar 5
77. Muscle Nerve 2009;38:1070-1073
78. Semin Respir Crit Care Med 2009;30:315�320, StatPearls
79. Biochemical Biophysical Research Communications 2007;363:1033�1037, Brain Dev 2023 Feb 14
80. Neurology 2009;73:1159-1161
81. Pflugers Arch 2010; Online Feb, Skelet Muscle 2020;10:32
82. Neuromuscular Disorders 2010; Online Mar
83. Journal of Clinical Investigation 2010; Online November
84. Am J Human Genet 2010; Online November, Front Neurol 2017;8:367
85. Neuromuscular Disorders 2011; Online April
86. Neuromuscular Disorders 2011;21:379�386, Neuromuscular Disorders 2011;21:387�395
87. Nature Genet 2011; Online November, Neurogenetics 2012; Feb 28, Acta Myol 2022;41:111-116
88. Neurology 2011;76:451-455
89. Semin Pediatr Neurol 2011;18:250-256, Int J Mol Sci 2021;22:11377
90. Am J Human Genet 2012; Online Jan
91. Nature Genetics 2012; Online April A, B
92. Acta Neuropathol 2012 Jul 3
93. Am J Human Genet 2012; Online July
94. Neuromuscul Disord 2012 Jul 9
95. Neuromuscul Disord 2012 Jul 23
96. American Journal of Human Genetics 2012;91;541�547
97. Eur J Hum Genet 2012 Oct 24
98. Neuromuscular Disorders 2012; Online November
99. Brain 2013; Online Jan
100. Am J Med Genet 2012;158A:772-778
101. J Neurol 2013; Online Jan
102. Brain 2013;136:282-293
103. Hum Mol Genet 2012;21:5484-5499
104. American Journal of Human Genetics 2013; Online Feb
105. Science 2013: Mar 21
106. Hum Mol Genet 2013;22:1746-1754
107. Neuromuscul Disord. 2013;23:540-548
108. Am J Human Genet 2013;93:29�41, Brain 2015; Online Feb, Genes (Basel) 2023;14:372
109. Hum Molec Genet 2013; Online August, Clin Genet 2020 Dec 22, J Genet 2023;102:18
110. Neurology 2013;Aug 23
111. Am J Human Genet 2103; Online Nov
112. Neuromuscular Disorders 2104; Online May, Eur J Med Genet 2023;66:104706
113. American J Human Genetics 2014; Online July, Hum Genome Var 2023;10:24
114. Pediatric Neurology 2014;51:192-197
115. J Clin Invest 2014;124: 4693-4708, Neurology 2018;91:e1690-e1694
116. Brain 2014; Online Sep
117. Amer J Human Genet 2014; Online Dec
118. Neuromuscular Disorders 2015; Online Feb
119. J Neurol 2015: May
120. JAMA Neurol 2015 May 4
121. Neuromuscul Disord 2015;25:488-492
122. Brain 2015; Online June
123. Ann Neurol 2015 Online September
124. Brain 2015; Online December, Neuromuscular Disorders 2017; Online Feb
125. J Neurol 2016 Jan 11
126. Eur J Hum Genet 2016 Jan 27
127. Am J Med Genet A 2016 Feb 11
128. Eur J Hum Genet 2015;23:883-886
129. Neurol Genet 2015;1:e33
130. Am J Human Genet 2016; Online May
131. Hum Mol Genet 2015;24:6146-6159
132. Neuromuscul Disord 2016 May
133. Am J Hum Genet 2016 Sep 28, Hum Mol Genet 2023 Mar 15
134. Brain 2016; Online November, J Hum Gen 2023;68:107-109
135. Muscle Nerve 2016;54:806-808
136. Am J Human Genet 2016; Online Dec
137. Am J Human Genet 2017; Online Feb
138. Neurology 2017 Feb 15, Am J Med Genet A 2021 Aug 20
139. Ann Neurol 2017; Online Feb, J Clin Neurol 2021;17:409-418
140. Neuromuscul Disord 2017 Feb
141. Hum Mol Genet 2017 Apr 13
142. Neuromuscular Disorders 2017; Online May
143. Nat Commun 2017 Jul 6
144. Eur J Neurol 2018 Mar 2
145. Am J Human Genet 2018; Online June
146. Hum Mol Genet 2018 Sep 12, Neuropathol Appl Neurobiol 2025;51:e70025
147. J Neurol 2018 Dec
148. Acta Neuropathol Commun 2019;7(1):3
149. Neuromuscular Disord 2019; March
150. Neuromuscul Disord 2017;27:742-746
151. Acta Neuropathol 2019;137:501-519, Ann Neurol 2019 Mar 22, Acta Neuropathol 2021 Sep 1, Hum Mutat 2022 Sep 18
152. Handb Clin Neurol 2019;162:435-448
153. Dis Model Mech 2019;12(8)
154. Acta Neuropathologica 2019;138:477�495
155. Nat Commun 2019;10:797, J Med Genet 2022 Apr 7
156. Acta Neuropathol Commun 2019;7:211, Am J Hum Genet 2020 Nov 17
157. Neuropathol Appl Neurobiol 2020 Aug 5, Cells 2021;10:2480
158. Respir Care 2020 Jun;65(6):807-819, J Clin Neurophysiol 2020;37:208-210, Physiol Rev 2025 Jul 11
159. Neuromuscul Disord 2015;25:567-576
160. Neurology 2020 Aug 13
161. Cell Death Differ 2020 Jul 13
162. Proc Natl Acad Sci U S A 2020 Aug:202003847
163. Neuromuscul Disord 2020 Dec 5
164. Neuromuscular Disorders 2019;29:97�107, medRxiv 2024:2024.10.04.24313542, Eur J Hum Genet 2025 Jun 14, Neuromuscul Disord 2026:62:106415
165. Nat Commun 2020;11:2699
166. Neuromuscul Disord 2020 Nov 30
167. Neurology 2020;95(24):e3406-e3411
168. FASEB J 2021;35:e21346
169. Hum Mutat 2018;39:1314-1337, Mol Genet Genomic Med 2020;8:e1387, Neuromuscul Disord 2025:55:106220
170. Neurotherapeutics 2018;15:885-899, Skelet Muscle 2020;10:32, Neuromuscul Disord 2021 Sep 17
171. Front Mol Neurosci 2020;13
172. Acta Neuropathol Commun 2021;9(1):155
173. Mol Genet Genomic Med 2021 Sep 16;e1804
174. Am J Med Genet A. 2021 Dec 4
175. Am J Med Genet A 2022 May 2
176. Neuromuscul Disord 2022 May 28
177. J Neuromuscul Dis 2022 Jun 20
178. Mol Genet Genomic Med 2022 Jun 27
179. Mar Biotechnol (NY) 2022 Sep 9
180. Semin Pediatr Neurol 2019;29:55-70
181. BMC Neurol 2015;15:223
182. Neurology 2022;99:e2223-e2233
183. J Med Genet 2023;60:48-56
184. Eur J Neurol 2022 Nov 27
185. Am J Med Genet C Semin Med Genet 2017;175:8-26
186. Cold Spring Harb Perspect Biol. 2022:a041246
187. Acta Neuropathol Commun 2021;9:155
188. J Med Genet 2021;58:602-608
189. Am J Med Genet A 2023 Jul 18
190. FASEB J 2024;38:e23400
191. Int J Mol Sci 2022;23:6274
192. medRxiv [Preprint] 2024 Feb 11
193. Neurol Genet 2021;7:e619
194. J Am Coll Cardiol 2010;55:1127-1135
195. J Am Heart Assoc 2022;11:e026494
196. Eur J Neurosci 2022;56:4214-4223
197. J Med Genet 2024 Dec 6
198. Orphanet J Rare Dis 2024;19:300
199. Genes (Basel) 2023;14:2174
200. Neuromuscul Disord 2025:49:105338
201. EMBO Mol Med 2025 May 23
202. J Neuromuscul Dis 2025 Jun 10
203. Mol Ther 2025 May 6
204. Cell Mol Biol Lett 2025;30:98
205. Brain Dev 2025;47:104437
206. Res Sq 2025 Sep 1
207. Front Genet 2025;16:1699834
208. Acta Neuropathol Commun 2023;11:20
209. Muscle Nerve 2024;69:55-63
210. J Hum Genet 2026 Mar 2
211. Hum Mol Genet 2026 May 11
212. Neurol Genet 2026;12:e200392
6/9/2026