|
|
BASIC CONCEPTS Acetylcholine receptors (AChRs) Disorders Structure Subunit mutations: α; β; ε; δ Neuromuscular junction (NMJ) Illustrations: A; B Presynaptic Postsynaptic Diagnostic tests ACQUIRED NMJ DISORDERS Botulism Myasthenia gravis Autoimmune myasthenia gravis Childhood MG Drug-induced MG Neonatal MG Transient AChR inactivation Arthrogryposis Ocular Anti-MuSK antibody positive Anti-AChR-antibody-negative Thymoma Domestic animals Myasthenic syndrome (Lambert-Eaton) Snake venom toxins  |
CONGENITAL & FAMILIAL NMJ DISORDERS
2
|




Myasthenic Syndrome (Lambert-Eaton; LEMS): Autoimmune
|
Antibodies Clinical features Electrophysiology Epidemiology Neoplasms Subgroups Treatment Also see Congenital LEMS syndrome 
|
LEMS: Increment after exercise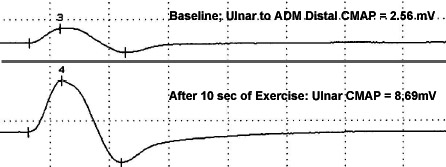 From M Al-Lozi |
- Epidemiology
17
- Prevalence: 1 in 100,000
- Sex ratio: Males slightly more common than Female
- Clinical features
- Onset
- Age
- Most > 40 years
- Range = 7 to 80 years
- Onset younger: No associated neoplasm
- Childhood: 5%
- Weakness (82%), especially legs
- Usually precedes cancer: > 80%
- Age
- Weakness
- Proximal > Distal
- Extremities
- Legs (98%)
- Arms (82%)
- Legs may be more severe than Arms
- Neck (30%)
- Respiratory (15%): Rarely severe; Later in disease course
- Bulbar: Dysphagia (22% to 56%); Dysarthria (Up to 80%)
- Improves with: Brief sustained exercise
- May worsen with: Sustained exercise; Heat or Fever
- Fatigability (33%)
- Extraocular muscle involvement
- Ptosis: Occasional (30% to 50%)
- Diplopia, Symptomatic (~ 40%): Transient
- Eye movement limitation: Rarely present at presentation or on examination
- Rule out: Concurrent myasthenia gravis
- Muscle pain: Occasional
- Sensory neuropathy: Distal; Symmetric
- Autonomic neuropathy (75%)
- Association stronger with cancer than with LEMS
- Dryness: Mouth > Eyes
- Impotence: Males
- Blurred vision
- Other (10% to 50%): Bladder; Obstipation; Hypohidrosis
- Tendon reflexes
- At rest: Diminished or absent (90%)
- Reappear after: Brief maximal voluntary contraction; Repeated tendon percussion
- Associated neurologic syndromes
- CNS
- Ataxia (5% to 56%)
- Encephalopathy
- More common in paraneoplastic LEMS
- Anti-Hu syndromes
- Weight loss (24%)
- "Viral" prodrome (34%)
- Cranial nerves: Usually normal
- CNS
- Course: Generally stable defect
- Exacerbation of LEMS by drugs: Occasional
- Neuromuscular junction blocking agents
- For surgery
- Botulinum toxin
- Antibiotics: Aminoglycoside; Fluoroquinolone, Ofloxacin
- Magnesium
- Ca++-channel blockers: Diltiazem, Verapamil
- IV contrast agents: Iodinated
- Varenicline (Chantix).
- Neuromuscular junction blocking agents
- Differences from Myasthenia Gravis
12
- LEMS never begins with ocular weakness
- LEMS usually has weakness in Legs > Arms
- Treatment: Often Beneficial
55
- Pharmacologic
- 3,4 Diaminopyridine
- 2 formulations
- 3,4-diaminopyridine phosphate (3,4-DAPP; Amifampridine phosphate)
- 3,4-diaminopyridine (3,4-DAP)
- Usual doses: 5 mg to 25 mg 3x or 4x per day
- Side effect: Seizures, especially doses > 100 mg/day
- 2 formulations
- ± Pyridostigmine
- 3,4 Diaminopyridine
- Immunosuppression
- Anti-T-cell drugs
- Plasma Exchange
- IV Ig
- Treatment of neoplasm
- Pharmacologic
- Onset
- LEMS Subgroups
5
- LEMS without neoplasm (SCLC)
- Epidemiology
- Male (50%) = Female (50%)
- Median age: 49.5 years
- 40% of LEMS
- Weakness
- More confined to proximal legs
- Many with some in arms
- Autonomic: Dry mouth (84%); Erectile dysfunction; Constipation
- Course: Slow progression with normal life expectancy
- Immunosuppression (Corticosteroids + ..)
- Improvement in most
- Continued immunosuppression needed in most
- Better prognosis with better clinical score at onset
- No correlation with: Anti-P/Q-VGCC antibody titers; Size of increment on RNS
- Lymphoma may be detected on follow-up (6%)
- Associated immunologic disorders
- Frequency: 27%
- Common types: Myasthenia gravis; Pernicious anemia; Thyroid;
Systemic lupus erythematosus; Celiac disease; Vitiligo; Diabetes
- HLA association: DR3-B8
- Epidemiology
- LEMS with neoplasm
- Epidemiology
- Male (70%) > Female (30%)
- Median age: 58 years
- Positive smoking history
- Frequency in patients with small cell lung cancer
- Clinical LEMS: 0.4% to 4%
- VGCC antibody: 8%
- Clinical
- Progression: More rapid 22
- Weakness
- Onset: Proximal legs
- Common progression: To arms & distal legs
- Respiratory failure more common
- Dysarthria
- Weight loss: More common
- Autonomic: Dry mouth; Impotence
- Shortened life expectancy: Probably related to neoplasm
- CNS: Ataxia more common (9%)
- Laboratory
- Associated immunologic disorders: 6%
- ESR: High
- Associated neoplasm
- Frequency: 25%
- Small cell lung cancer
- Common
- May be less malignant
23
- Long survival more common
- Average suvival with LEMS 2x longer: 18 months to 4 years vs 9 months
- No clear increased survival with other paraneoplastic syndromes
- ? Merkel cell carcinoma
- Epidemiology
- LEMS without anti-(VGCC) antibodies
13
- Frequency: 15% of LEMS patients
- Clinical features: Similar to antibody positive group
- Less associated small cell lung cancer: 12% vs 60% with antibodies
- Survival when SCLC present: Shorter that antibody-positive patients
- Serum IgG reduces quantal content of end plate potentials
- LEMS without neoplasm (SCLC)
- Pathophysiology: Presynaptic disorder
- Reduced numbers of P/Q Ca++ channels on presynaptic terminals
- Ultrastructure: Active zone particles on presynaptic terminals reduced
- Physiology
- Reduced K+ stimulated Ca++ flux into presynaptic terminals ®
- Reduced Ca++ dependent quantal ACh release
- Serum Antibodies
- IgG vs Voltage Gated Calcium Channels (VGCC)
- Location: Serum ± CSF
- Frequency in LEMS
- Overall: 85%
- LEMS + Cancer: 98% Ab+
- LEMS without cancer: 80% Ab+
- More common: With decrement at slow stimulation rates
- Other positive testing
- CNS disorders: Cerebellar disorders with neoplasm
- VGCC antibody frequency: 40%; More common than Hu
- Neoplasm type: Lung most common, especially small cell
- Intrathecal synthesis: Some patients
- Response of cerebellar syndrome to treatment: Poor
- LEMS clinical & electrodiagnostic features: Often not present
- Small cell lung cancer
- False positives
- Hypergammaglobulinemia
- Chronic liver disorders
- Infections
- Normal population: < 3%
- CNS disorders: Cerebellar disorders with neoplasm
- Target antigens
- P/Q-type Ca++ channel (85%): α1A subunit; Especially domains II & IV
- N-type Ca++ channel (35%): ? α1B subunit
- Other associated serum antibodies
- Glutamic acid decarboxylase (24%)
- Thyroid (21%)
- Parietal cell (14%)
- Hu (3%)
- Muscle nicotinic AChR (10%)
- Synaptotagmin (20%)
- M1 muscarinic AChRs
- SOX1
 antibodies
27
antibodies
27
- Present
- Small cell lung neoplasm (63%)
- LEMS patients with associated Small cell lung neoplasm (64%)
- IgG vs Voltage Gated Calcium Channels (50%)
- Not present: LEMS patients without SCLC (0%)
- Cerebellar staining pattern: Nuclei of Bergmann glia (AGNA)
- Other associations
- Hu Ab (32%)
- Small cell lung cancer only (22%);
- Voltage-gated K+ channel Ab
- Present
- IgG vs Voltage Gated Calcium Channels (VGCC)
- Electrophysiology
- Repetitive nerve stimulation
- Increment
- After rapid (50 Hz) RNS, or
- Sustained muscle contraction
- Prolonged by cooling
- Muscles with most increment after 10 sec maximal voluntary contracture
- Most specific for LEMS
- > 60% to 100% in several muscles
- > 400% in one muscle
- Muscles: Abductor digiti minimi; Abductor policis brevis; Anconeus
- Decrement on slow (5 Hz) RNS: Especially in small hand muscles
- Increment
- Small CMAP amplitude
- Microphysiology
- Ca++ channel blockade: P most common; Q often; N minority of cases
- Reduced quantal acetylcholine release from presynaptic nerve terminal
- Repetitive nerve stimulation
- Tumor
- Clinical: See LEMS subgroups
- Frequency of association with neoplasm
- Overall: 50% to 60%
- With risk factors: Up to 100%
- Risk factors: Smoking; Increasing Age (> 50 years)
- Associated neoplasms
- Small Cell Lung Cancer
- Most frequent associated neoplasm
- Clincial LEMS: 3% to 5% of patients with SCLC
- Serum IgG binding to VGCC: 10%; 8% to P-type calcium channel
- Almost all SCLC with LEMS have smoking history
- Lymphoproliferative: Reticulum-cell sarcoma; T-cell leukemia; Lymphoma; Castleman's disease
- Other possible associations: Non-small cell lung; Prostate; Thymoma; Merkel cell carcinoma
- Small Cell Lung Cancer
- Timing of neoplasm association
- Usual (80% to 90%): LEMS onset 0.5 to 5 years before neoplasm detected
- Neoplasm only found in follow-up
- Frequency: 10%
- Most commonly: < 1 year; Range 1 to 41 months
- Work-up: Chest CT or MRI
- If no neoplasm found at initial evaluation: 3 months, then q 1 year x 5 years later
- Chest X-ray: Not sensitive
- FDG-PET: May identify additional cases
- Immunization with extracellular region (S5-S6 linker) of domain III of P/Q Ca++ channel 1
CONGENITAL MYASTHENIC SYNDROMES (CMS) 2
|
Acetylcholine receptors (AChRs) Kinetic abnormalities Mutations Differential diagnosis Clinical features Cell & Molecular Treatments |
CONGENITAL MG SYNDROMES: Clinical features
- Genetics: Inheritance
- Recessive: Most congenital myasthenic disorders
- Dominant: Slow AChR Channel syndromes
- Clinical
- Onset
- Early: Most congenital MG; Often infancy or childhood
- Later
- Slow-channel syndrome: Some
- Familial limb-Girdle myasthenia (Dok-7)
- Rapsyn: Some
- AChR (Non-epsilon): Low expressor
- Weakness
- Ocular; Bulbar; Respiratory; Generalized
- Wrist & finger extensors: Older patients with
- Limb-girdle & Axial: Dok7, GFPT1, DPAGT1, ALG2, ALG14, Rapsyn, ColQ
- Stridor & vocal cord paralysis, Infancy: Dok-7 > ColQ, MuSK
- Milder ocular involvement
- Pupil
- Delayed light reflexes: Endplate AChE deficiency (ColQ)
- Persistently constricted: Laminin-β2
- Variable course
- Fatigue & Fluctuation of signs: Especially with MG + episodic apnea (ChAT)
- Episodic apnea in childhood
- MG + episodic apnea (ChAT)
- AChR syndromes
- Rapsyn mutations
- Sudden apneic episodes after fever or stress: ChAT, Rapsyn, Na+ channel
- Tendon reflexes reduced
- Arthrogryposis (Contractures)
- Recurrent congenital MG: Maternal antibodies vs fetal AChRs
- Multiple pterygium syndrome (Escobar): AChR δ or γ subunit mutations
- Rapsyn
- ChAT
- ECEL1
- ZC4H2
- ZBTB42
- Clinical: Other
- Progression
- Usual: Slow or none in adolescence or adulthood
- More progression
- CMG: Treatments
- Anti-AChE medications
- Mechanism: Blocks ACh degradation by AChE
- AChR deficiency syndromes
- ChAT deficiency
- Not effective: AChE deficiency
- Refractory to, or worsened by, AChE inhibitors: ColQ, Dok-7, MuSK, Agrin, LRP4, Plectin, Laminin β2, Rapsyn
- 3,4-Diaminopyridine
- Salbutamol (Albuterol)
- Mechanisms: Stimulates β2-adrenergic receptors; May activate MuSK; Effects on AChR clustering
- Disorders: Dok-7, AChE deficiency, Laminin β2, LRP4, COL13A1, Rapsyn, Agrin
- Ephedrine
- Mechanism: Stimulates β2-adrenergic receptors; May activate MuSK
- Disorders: Dok-7, AChE deficiency, Agrin, Laminin β2
- Fluoxitine
- Mechanism of action: AChR channels
- Open time reduced
- Reduced opening frequency: Stabilizes AChR desensitized state
- Disorder: Slow-channel syndromes
- Mechanism of action: AChR channels
- Quinidine
- Mechanism of action: AChR channel open time reduced
- Disorder: Slow-channel syndromes
- ? Corticosteroids
- Anti-AChE medications
- Laboratory
- Antibodies vs AChRs: Absent in hereditary myasthenic syndromes
- Tensilon test: Often positive
- Negative: Endplate AChE deficiency
- Electrophysiology
- Decrement on Repetitive nerve stimulation
- Common: At 2Hz stimulation
- Absent: CMG with episodic apnea when asymptomatic
- Absent at rest, but elicted by 10 Hz stimulation: ChAT deficiency
- > 50% decrease of CMAP amplitude after 10 Hz stimulation for 5 min & slow recovery over 5–10 min: ChAT
- Not corrected by edrophonium: Endplate AChE deficiency
- Repetitive CMAP to single stimulus
- Endplate AChE deficiency (ColQ)
- Slow-channel syndrome: Dominant AChR subunit mutations
- PURA-CMS
- Patients taking high doses of AChE inhibitors
- Decrement on Repetitive nerve stimulation
- Morphology
- NMJ
- No IgG or complement at NMJs
- Small endplate regions, Multiple: Slow-channel syndrome; AChR deficiency syndromes
- Absent AChE staining: Endplate AChE deficiency
- Reduced numbers of junctional folds in post-synaptic membrane
- Molecular evaluation of NMJs: AChE, AChR, Agrin, B2-laminin, Utrophin, Rapsyn
- Muscle
- NMJ
CONGENITAL & FAMILIAL NMJ DISORDERS 2: Molecular differential diagnosis
General: Syndromes differentiated by anatomic location of mutated protein
- Presynaptic
- Defects in evoked release of acetylcholine (ACh) quanta or ACh resynthesis (ChAT mutations)
- Frequency: 8%
- Synaptic basal lamina
- Defects caused by mutations in
- Collagen tail of AChE: Frequency 16%
- Laminin β2
- Defects caused by mutations in
- Many caused by mutations in subunits of AChRs
- Other: Syndromes caused by other post-synaptic molecules
- Frequency: 76%
- CMSTA1: GFPT1; 2p13
- CMSTA2: DPAGT1; 11q23
- CMSTA3: ALG2; 9q22
- Familial MG: ALG14; 1p21
CONGENITAL MG SYNDROMES: General types of AChR Kinetic abnormalities & Other disease features
- Slow channel syndromes
- Inheritance: Dominant; Occasionally recessive
- Mutations: AChR subunits
- Channel physiology: Opened for prolonged time
- Endplate currents: Slow decay
- Channel opening events: Prolonged
- Open states: Stabilized
- Closed states: Destabilized
- Mechanisms of abnormality
- AChR Affinity for ACh: Increased
- AChR channel opening rate (β): Increased
- AChR channel closing rate (α): Decreased
- Clinical & Pathology: Slow channel syndromes
- Congenital myasthenic syndromes, Fast channel
33
● CMS1B: Acetylcholine receptor α subunit (CHRNA1); Chromosome 2q31.1; Recessive
● CMS2B: Acetylcholine receptor β subunit (CHRNB); Chromosome 17p13.1; Recessive
● CMS3B: Acetylcholine receptor δ subunit (CHRND); Chromosome 2q37.1; Recessive
● CMS4B: Acetylcholine receptor ε subunit (CHRNE); Chromosome 17p13.2; Recessive
- Genetics
- Inheritance: Recessive
- Mutation effects: Loss of function
- Consequences of fast channel mutation unmasked by null mutation in other allele
- CHRNA1: Allelic disorders
- AChR channel physiology
- Endplate currents: Rapid decay
- Channel opening events: Brief
- Open states: Destabilized
- Closed states: Stabilized
- Mechanisms of abnormality
- Specific channel abnormalities: Brief, or reduced, channel activation due to several mechanisms
- Decreased affinity for ACh
- εP121L mutation: Common
- Mutation effect
- Reduced affinity for ACh in AChR open state
- Number of AChRs at NMJs: Often normal
- Location
- Extracellular domain of ε subunit
- Near ACh ligand binding site formed by α & ε subunits
- Physiology: Slowed channel opening
- Clinical phenotype: Moderately severe MG
- Mutation effect
- εD175N mutation
- Mutation effect: Reduced affinity for ACh in AChR closed state; Impaired gating efficiency
- Location: Near ACh ligand binding site formed by α & ε subunits
- δE59K mutation
- Mutation effect: Reduced affinity of AChR for ACh
- Location: Near ACh ligand binding site formed by α & δ subunits
- Clinical phenotype: Multiple congenital joint contractures; Neonatal respiratory distress
- εP121L mutation: Common
- Abnormal channel gating mechanism
- εTrp55Arg mutation
- Mutation effects: Gating abnormal & Reduced ACh affinity
- Abnormal isomerization of AChR from closed to open state
- Slow opening rate
- Fast closing rat
- Reduced open channel probability
- Clinical phenotype: Severe; Congenital; Early death
- Mutation effects: Gating abnormal & Reduced ACh affinity
- εN182Y mutation
- Mutation effect: Increases ACh affinity for the resting closed state
- Clinical phenotype: Moderately severe MG
- αV285I mutation
- Location: M3 transmembrane domain of α subunit
- Mutation effect: Impaired gating efficiency
- Clinical phenotype: Mild MG
- εR311W mutation
- εAla411Pro mutation
- Heterogeneous gating kinetics
- Location: Amphipathic helix of the long cytoplasmic loop
- Clinical phenotype: Mild to moderate MG
- εTrp55Arg mutation
- Abnormal mode-switching kinetics: ε1254ins18 mutation in cytoplasmic loop of ε subunit
- Reduced probability of channel opening after ACh binding
- αV132L mutation
15
- Impaired ACh binding to AChRs in the resting closed state
- Decreasing binding affinity for the second binding step 30-fold
- Gating efficiency impaired only 2-fold: Similar mutation in δAChR subunit impairs gating
- Location: Cys-loop domain of α subunit
- Clinical phenotype: Most severe fast channel MG syndrome
- Generalized weakness
- Respiratory failure
- αV132L mutation
15
- Fast channel syndrome with arthrogryposis: δ subunit
- Reduced expression of AChRs with fast channel properties
- Decreased affinity for ACh
- Clinical: General
- Onset: Birth; Respiratory or Feeding problems; Fetal movements reduced
- Episodic: Commonly develop life threatening episodes of apnea in childhood
- Weakness: Proximal > Distal; Face & Neck
- Ophthalmoplegia: Common
- Treatments: Partial response to cholinesterase inhibitors & 3,4-diaminopyridine
- Clinical correlations
- Mild disease: Abnormal gating efficiency
- Moderate severity: Unstable channel kinetics
- Severe disease: Abnormal ACh affinity ± Impaired gating efficiency
- Treatment
- 3,4-diaminopyridine
- Best response when normal numbers of AChRs at NMJs
- Pathology
- May be Normal
- Reduced numbers of AChRs at NMJs
- Multiple small endplate regions
- Genetics
Episodic Apnea
CMS6: Congenital MG with Episodic apnea
● Choline acetyltransferase (ChAT)
- Nosology: Other nammes
- Defect in ACh resynthesis or packaging
- Familial Infantile Myasthenia
- Gene mutations
- Often missense
- Occasionally 1 mutation is frame shifting
- Most patients compound heterozygotes
- Turkish families with homozygous I336T 18
- ChAT protein
- ChAT protein
 : Normal features
: Normal features
- Location: Presynaptic nerve terminals
- Function: Facilitates transfer of acetate to choline; Produces acetylcholine
- External link: Williams
- Mutation effects on ChAT protein
- Kinetic disorders
- Cause loss of function of ChAT
- Often produce altered affinity of ChAT for AcCoA
- Protein expression: Often reduced, but not absent
- Kinetic disorders
- ChAT protein
- Clinical features: Variable
- Onset
- Birth, or Early childhood
- Hypotonia
- Weakness: Bulbar & Respiratory
- Gradual improvement
- Weakness: Episodic rapid exacerbations
- Onset: Infancy or Childhood
- Ptosis: Fluctuating
- Crises
- Signs: Respiratory failure; Episodic sudden apnea; Weakness; Bulbar dysfunction
- Precipitated by: Fever; Exertion; Excitement
- Gradual improvement: Respiratory impairment may persist for weeks
- Partial or complete recovery after each episode
- Between episodes
- Normal, or
- Moderate myasthenic symptoms: Ptosis; Fatigability
- Course
- Improvement with age: Fewer exacerbations
- Occasional sudden death
- Tendon reflexes: Normal
- Other
- Muscle: No myopathy; Normal muscle bulk
- Autonomic: Normal
- CNS: Normal
- Treatment
- AChE inhibitors: Prophylactic pyridostigmine
- ? 3,4 diaminopyridine
- Availability of emergency ventilatory apparatus (Bag)
- Apnea monitor
- Avoid: Aminoglycoside antibiotics; Quinidine
- Onset
- Electrophysiology
10
- Decrement with repetitive nerve stimulation
- Variably present
- Often not present in normal strength, rested muscles
- More common during attacks
- On 2 to 3 Hz RNS: Only in weak muscles
- Decrement increased after
- Exercise
- 10 Hz stimulation of several minutes
- Slow recovery of CMAP to normal amplitude after RNS finished
- Corrected by
- Rest: Improves slowly over > 2 minutes
- Edrophonium
- Variably present
- Single stimulus evokes single CMAP
- CMAP amplitude: Normal at rest; Reduced with 10 Hz RNS for 5 minutes
- MEPPs
- Normal in resting muscle
- Decreased Amplitude after 10 Hz RNS for 5 min: Persists for 10 to 15 minutes
- End-plate potential (EPP): Quantal size Reduced; Quantal content normal
- Decrement with repetitive nerve stimulation
- Pathology: Presynaptic disorder
- Probable mechanism: Reduced acetylcholine resynthesis or repackaging in presynaptic terminal
- Synaptic vesicles
- Rested muscle: Small synaptic vesicles
- Stimulation: Increase or no change in size
- Post-synaptic: Normal number of AChRs; Normal post-synaptic morphology
CMS21: Congenital MG with Episodic apnea
● Solute carrier family 18 (Vesicular acetylcholine), Member 3 (SLC18A3; VAChT)
- Epidemiology: 4 patients; Yemeni Jewish; Philipino & Turkish
- Genetics
- Mutations: c.557G>C, p.(Gly186Ala); Deletion; c.1192G.C, p.(Asp398His); c.1078G>C, p.Gly360Arg
- Gene is exon in 1st intron of ChAT gene
- More severe disease: Gly360Arg
- SLC18A3 protein
- Presynaptic
- Uptake of acetylcholine from cytoplasm into presynaptic vesicles
- Clinical
- Onset age: Congenital
- Eye
- Ptosis
- Ophthalmoplegia
- Weakness
- Proximal
- Arms & Legs
- Face, mild
- Dysphagia
- Respiratory
- Fixed: More severe syndromes
- Episodic: Cyanosis or Apnea
- Fatigue: Worse with cold water
- Tendon reflexes: Present
- Skeletal
- Contractures
- Arthrogryposis: More severe patients
- Cognitive defects: Mild, 1 patient
- Treatment: Pyridostigmine improves fatigue & strength
- Laboratory
- Repetitive nerve stimulation
- 3 to 5 Hz Decrement
- Postactivation exhaustion
- Unmasked by isometric contraction
- Cardiac: Abnormal LV function
- Repetitive nerve stimulation
Paucity of Synaptic Vesicles and Reduced Quantal Release: Congenital
- Epidemiology: 1 patient reported
- Clinical features
- Onset: Birth
- Weakness
- Bulbar & Limb
- Ptosis & Face
- Extraocular movements: Limited in some patients
- Fatigability
- Treatment: AChE inhibitors
- Electrophysiology
- Repetitive nerve stimulation: Decrement
- Endplate potential (EPP): Quantal content reduced to 20% of normal
- CMAP amplitude: Normal
- Synaptic morphology
- Presynaptic nerve terminal
- Synaptic Vesicles: Reduced in number by 80%
- Terminal size: Normal
- Post-synaptic: Normal folds; Normal number of AChRs
- Presynaptic nerve terminal
- Possible defect: Reduced synthesis or axonal transport of vesicle precursors
CMS5: Endplate Acetylcholinesterase (AChE) Deficiency
● Collagenic Tail of Endplate Ecetylcholinesterase (ColQ)
- Epidemiology
- Genetics
- >20 Different mutations described
- Mutation types
- Missense mutations (P59Q; D342E): Milder disease
- Point mutations → Stop codons: Severe disease
- Mutations
- G240X mutation: Common in Palestinian Arabs
- Homozygous missense (P59Q; R410W; Tyr430Ser): May have Teen onset
- Functional effects of mutations differ
- N-terminal (P59Q) or PRAD region (107del215)
- Reduced Binding of ColQ to catalytic subunit of AChE
- More distal mutations
- Insertion of ColQ into basal lamina: Reduced
- Formation of asymmetric forms of AChE: Reduced
- N-terminal (P59Q) or PRAD region (107del215)
- Protein: ColQ
- Collagen
- Collagen tail of asymmetric acetylcholinesterase
- Proline-rich attachment domain (PRAD) of ColQ binds tetramer of AChE subunits
- Produces asymmetric AChE: Concentrated at NMJ
- ColQ tail anchors catalytic AChE subunits
to basal lamina
- AChE stabilization depends on binding to
- Perlecan-Dystroglycan
- MuSK: Responsible for AChE accumulation at synaptic cleft2
- Tail formed by triple helix association of 3 collagen-like strands (ColQ)
- AChE
- Several isoforms
- Vary in C-terminal domain
- AChET variant at NMJ & Brain
- Located in synaptic cleft: Hydrolyzes ACh
- See: Anti-AChE antibodies
- AChE stabilization depends on binding to
Acetylcholinesterase
A12, Asymmetric - Clinical features
- Onset
- Birth to 2 years: Variable even with same mutations
- Weakness: Generalized; Respiratory in some patients
- Weakness
- Slow motor milestones
- Diffuse: Respiratory, Extraocular, Facial, Proximal trunk & Limb
- Symmetric
- Severely disabling: Most patients
- Tendon reflexes: Reduced or Normal
- Slow pupillary response to light: Some patients
- Scoliosis: Older patients
- Tensilon test: May make patient worse
- Treatment
- Ephedrine: 50 mg qid 24
- Salbutamol: 2mg tid
- No response to AChE inhibitors
- Course: May be progressive, stable or improve
- Onset
- Electrophysiology
- Decrement at 2 Hz RNS
- In all muscles
- Not corrected by edrophonium
- Present in most patients: May develop during childhood
- Repetitive CMAP response to single stimulus (80%)
- Especially in well rested muscle
- Present in all patients: May be absent during infancy
- Repetitive CMAP is smaller amplitude & decrements faster than first CMAP
- Microphysiology: Small EPP due to reduced number of immediately releasable quanta
- No effect of AChE inhibitors on electrophysiological abnormalities
- Decrement at 2 Hz RNS
- Muscle Pathology: Endplate myopathy
- AChE deficiency at neuromuscular junctions: Absent asymmetric forms
- Small presynaptic motor nerve terminals
- Simplified post-synaptic folds at some NMJs
- Number of AChRs reduced
- Schwann cell processes extend into the synaptic cleft
- AChE: Partial AChE deficiency
- Onset: ~ 6 years
- Milder weakness
- Disabling during 2nd decade
- AChE Variant
- Mild symptoms in childhood
- Worsening in 5th decade with severe respiratory insufficiency
- Mutations
- Splicing (IVS1-1GtoA): Affects exon encoding proline-rich attachment domain (PRAD)
- R236X
- Sphynx & Devon Rex Cats
42
- ColQ mutations: Homozygous; c.1190G>A (C397Y); Missense
- Clinical: Weakness; Fatigability
Congenital Myasthenic Syndrome 30
● Laminin β2 (LAMB2)
- Epidemiology: Single patient
- Genetics
- Heterozygous mutations: 1478delG, 4804delC
- Allelic with
- Pierson syndrome
(Congenital nephrosis & Ocular defects)
- Nephrotic syndrome, type 5
- Pierson syndrome
- LAMB2 protein
- Laminins: Heterotrimeric glycoproteins containing &alpha, &beta & γ chains
- Location: Basal lamina
- NMJ laminins: Laminin-4, -9 & -11; All contain β2 chain
- LAMB2 deficient mice: Defective neuromuscular synapses; Schwann cells intrude in synaptic cleft
- Clinical
- Neonatal: Episodes of respiratory distress
- Eyes
- Pupils: Persistently constricted
- Ptosis
- EOM: Limited
- Myopia: Impaired visual acuity
- Fundus: Hypoplastic macula
- Motor
- Developoment: Delayed
- Weakness: Proximal
- Scoliosis
- Treatments
- Ephedrine
- AChE inhibitor: Increased weakness
- Renal transplant
- Laboratory
- Nephrotic syndrome: Proteinuria
- Repetitive nerve stimulation (3 Hz): Decrement
- Endplate physiology
- Reduced quantal content of end plate potentials (EPPs) evoked by nerve stimulation at 1 Hz
- MEPP frequency & amplitudes: Reduced
- Muscle biopsy
- General morphology: Non-specific changes
- Angular muscle fibers: Scattered
- Type I muscle fiber predominance
- LAMB2 staining: Absent
- Neuromuscular junctions (NMJs)
- Presynaptic
- Axon terminals: Small size
- Encasement of nerve endings by Schwann cells
- Synaptic vesicles: Normal to reduced numbers
- Primary synaptic cleft
- Severe widening; Invasion by Schwann cell processes
- Endplate acetylcholinesterase: Normal
- Presynaptic
- Postsynaptic folds: Simplified
- Endplate area: Reduced
- General morphology: Non-specific changes
Congenital Myasthenic Syndrome 48
● Laminin α5 (LAMA5)
- Epidemiology: 1 or 2 patients
- Genetics
- LAMA5 mutation: Homozygous; Arg2659Trp
- Other mutation: LAMA1, Homozygous, Asp210Asn
- LAMA5 protein
- Laminin
- Present at neuromuscular junction
- Role in presynaptic differentiation
- Clinical
- Onset age: Birth
- Hypotonia
- Weakness
- Respiratory: Mechanical ventilation
- Feeding: G-tube
- Eyes: EOM limitation; Ptosis
- Cranial: Face; Tongue; Soft palate
- Neck flexion
- Limbs: Proximal
- Tendon reflexes: Reduced
- Dysmorphism: Elongated face; Prosis; High arched palate
- Systemic
- GI: Inflammatory bowel disease
- Myopia
- Knee contractures
- Treatment: Pyridostigmine sulfate + 3,4-DAP
- Laboratory
- Repetitive nerve stimulation
- 2Hz: 55% decrement
- 30 s of maximal muscle activation: LEMS-like
- 250% increment
- Microphysiology
- EPP quantal content: Reduced
- MEPPs: Normal
- Anconeus muscle biopsy
- Type 1 muscle fiber predominance
- Normal: AChE, Laminin α-5, AChRs
- NMJ Ultrastructure
- Post-synaptic folding folding: Normal or increased
- Synaptic vesicile density: Moderately reduced
- Repetitive nerve stimulation
Slow Acetylcholine Receptor (AChR) Channel Syndromes (SCCMS; CMS3)
● Acetylcholine receptor α subunit (CHRNA1)
● Acetylcholine receptor β subunit (CHRNB)
● Acetylcholine receptor δ subunit (CHRND)
● Acetylcholine receptor ε subunit (CHRNE)
- Epidemiology
- Genetics: Multiple mutations identified
- Mutations: General
- Types: All missense
- > 25 identified
- Mutation features
- Locations
- Common: M2 (2nd) ion pore transmembrane domain of AChR subunits (60%)
- Other: 1st & 4th transmembrane domains; Extracellular (M2-M3) linker; Near AChR binding site
- Different functional effects
- Slow AChR ion channel closure
- Abnormal reopening of AChR
- Enhanced agonist binding affinity of AChR
- Increased AChR channel openings during ACh occupancy
- Decreased desensitization of mutant AChR by ACh
- Reduced number & density of AChRs at NMJs
- Clinical correlations
- Most severe with missense introduction of phenylalanine:
εL269F
;
αV249F
- Generalized weakness may be present
- Most severe with missense introduction of phenylalanine:
εL269F
- Other SCS mutations: αT254I;
βL263M
;
εT264P;
βV266M
;
δS268F
- Locations
- α subunit (CHRNA1) mutations
- CMS1A: Myasthenic syndrome, congenital, slow-channel
- M1 transmembrane domain: αN217K
- Clinical
- Moderate weakness: Forearm & Face
- Fatigability
- Mutation effects
- Slows rate of channel closing
- Allows multiple reopenings per activation episode
- Other M1 mutation: βV229F
- Clinical
- Extracellular, near ACh binding site: αG153S
- Decreased rate of dissociation of ACh from AChR
- Repeated openings with normal open time
- Mildest clinical syndrome
- Extracellular: αV156M
- ? Stabilizes open state
- M2 domain: αV249F: Not facing channel lumen
- Increased channel openings in absence of ACh
- Increased ACh affinity for AChR in closed state
- Prolonged or repeated opening in presence of ACh
- Enhanced steady-state desensitization
- Defects produce cationic overload & degeneration of junctional folds
- Mutation in corresponding valine to phenylalanine in ε subunit (V259F)
- Causes slow-channel syndrome
- Extracellular between M2 & M3: αS269I
- ? Alters coupling between ACh binding & channel gating
- CHRNA1: Allelic disorders
- CMS1A: Myasthenic syndrome, congenital, slow-channel
- β subunit (CHRNB)
mutations
- CMS2A: Myasthenic syndrome, congenital, 2A, slow-channel
- Allelic disorders
- βL262M mutation: More severe weakness
- Near channel gate
- Increased folding of postsynaptic membrane
- Increased number of AChRs
- CMS2C: AChR deficiency
- Epidemiology: 1 family
- Mutations: 9-BP DEL, NT1276; EX8DEL
- βL262M mutation: More severe weakness
- CMS2A: Myasthenic syndrome, congenital, 2A, slow-channel
- δ subunit (CHRND)
mutations
- CMS3A: Myasthenic syndrome, congenital, slow-channel 3A (Ser268Phe)
- Allelic disorders
- CMS3B: Myasthenic syndrome, fast-channel congenital
- CMS3C: AChR deficiency
- δS268F mutation 4
- Widening of synaptic cleft
- Accumulation of debris in synaptic cleft
- Normal post-synaptic morphology
- AChR #: Mildly reduced
- Multiple pterygium syndrome, lethal type
- CMS3B: Myasthenic syndrome, fast-channel congenital
- CMS3A: Myasthenic syndrome, congenital, slow-channel 3A (Ser268Phe)
- ε subunit (CHRNE)
mutations
- CMS4A: Myasthenic syndrome, congenital, slow-channel
- Allelic disorders
- CMS4B: Myasthenic syndrome, fast-channel congenital
- CMS4C: AChR deficiency
- CMS4B: Myasthenic syndrome, fast-channel congenital
- εLeu221Phe mutation
- Inheritance: Dominant: Variable penetrance
- Physiology: Prolonged AChR ion channel activations
- Mutation location: M1 domain of AChR molecule
- Disease severity: Mild; Congenital
- εV259F mutation
- Clinical: Late walking; Nasal speech; Ptosis; Proximal & face weakness
- Electrophysiology
- RNS decrement (on 2nd study)
- Jitter: Abnormal
- No repetitive CMAP
- Muscle pathology
- Endplate myopathy
- Tubulofilamentous inclusion bodies
- Mutation in corresponding valine to phenylalanine in α subunit (V249F)
- Causes severe slow-channel syndrome
- εSer278Del + εArg217Leu
- Inheritance: Recessive
- Physiology: Prolonged AChR ion channel activations
- Mutation location: Extracellular region of AChR molecule
- Disease severity: Mild
- CMS4A: Myasthenic syndrome, congenital, slow-channel
- Mutations: General
- Pathophysiology: General
- AChR Channel changes
- Prolonged opening episodes
- Slow decay of miniature endplate potential
- Caused by: Increased affinity of AChR for acetylcholine; Increased gating efficiency of channel
- Spontaneous channel openings of unliganded AChR
- Prolonged opening episodes
- Activated by exposure to synaptic ACh & Serum choline
- Cationic (Ca++) overload of junctional sarcoplasm
- Causes endplate myopathy with loss of junctional AChR
- Depolarization block: Due to summation of prolonged endplate potentials
- Endplate myopathy
- Focal activation of caspases 3 & 9 at NMJs 20
- AChR Channel changes
- Clinical features
- General: Variable phenotypes
- Mild: Later onset
- Severe: Infantile onset
- Onset: Infant to 7th decade; Typical 0 to 3 years
- Cranial nerves
- Often spared in milder cases
- Ptosis
- Ophthalmoplegia: More severe cases
- Facial weakness: Especially eye closure
- Weakness & Wasting
- Fatigability
- Spinal deformities: With disease progression
- Progression: Slow; More with earlier onset
- Treatments
- Quinidine Sulfate (60%)
- Dose: 200 mg 3 or 4 times per day; po
- Serum concentrations 0.7-2.4 μg/ml
- Mechanism of action: Blocker of open channel AChRs
- Clinical response: Gradual improvement
- Fluoxetine (100%) (Open AChR channel blocker): 80 mg to 160 mg/day
- Salbutamol (β2-adrenergic receptor agonist): 2 mg to 4 mg tid
- Ephedrine
- Chloroquine
- AChE inhibitors: No long term response; 50% with some initial response
- Quinidine Sulfate (60%)
- General: Variable phenotypes
- Electrophysiology
- Repetitive nerve stimulation (75%): Decrement in weak muscles
- Repetitive CMAP response to single stimulus
- More with: Longer AChR open times (> 4x normal)
- Exacerbated by: Edrophonium
- Jitter: abnormal
- Postsynaptic recordings
- Disorders due to: Slow closure of AChR ion channel
- Miniature endplate currents: Prolonged & Biexponentially decay
- Staircase summation
- Depolarization block during subtetanic stimulation
- Single channel recordings
- Dual population of AChR channels
- Normal channel: Normal opening episodes
- Mutant channel: Prolonged opening episodes
- Mutant chanels open even in absence of ACh
- Dual population of AChR channels
- Pathology
- Type I muscle fiber predominance
- Endplate myopathy
- Ca++ increased in muscle cytoplasm near NMJ
- Autophagic vacuoles: Regions neighboring NMJ
- Junctional folds: Severe damage; Similar to Dok-7 mutations
- Apoptosis: Nuclei at NMJs
- Reduced nerve terminal area
- Some patients with
- Tubular aggregates
- Tubulofilamentous inclusions
- Also see: Acquired slow channel syndrome
AChR Deficiency and Short Channel Open Time: Altered Mode-switching Kinetics
● Acetylcholine receptor ε subunit (CHRNE)
- Mutations: Heterozygous
- εC128S
- Prevents formation of conserved disulfide group in extracellular domain
- Arrests subunit assembly before formation of AChR pentamers
- ε1254ins18
- Reduces AChR expression
- Mutation adds new modes of gating to normal high efficiency
- Effects of mutation on channel function
- Slow opening of channels
- Rapid closing of channels
- εC128S
- Clinical features
- Onset < 2 years
- May be More common in males
- Weakness: Extraocular, Facial & Generalized
- Course: Benign; Weakness persists into adulthood
- Treatment: AChE Inhibitors; 3,4 Diaminopyridine
- Electrophysiology
- Decrement in facial muscles
- No exhaustion phenomenon
- Short open time of AChR channel
- Muscle
- Postsynaptic reduction in number of of AChRs
- Elongated neuromuscular junctions
- Simplified post synaptic folds
- Compensatory increase in expression of γ AChR subunit
- AChR function: Heterozygous εC128S & ε1254ins18
- Some channels with short open times
- Other channels with long open times: Probably AChRs containing γ, instead of ε subunit
- Mode switching kinetics
- 2 new AChR modes, both with inefficient gate
- In both modes channel opens more slowly and closes more rapidly than normal
- Electrically silent during synaptic response to ACh
AChR conversion into Chloride conductance at Positive potentials 50
● Acetylcholine receptor α subunit (CHRNA1)
- Epidemiology: 1 patient
- Genetics
- Mutation: Heterozygous; α1Leu251Arg
- Clinical
- Onset age: Infancy
- Feeding difficulties, Dribbling & Reduced facial expression,
- Motor milestones: Delay
- Eyes: Ptosis, bilateral & Fatigable; EOM limited
- Weakness: Face; Neck flexor; Limbs normal
- Fatigue: With exercise
- Laboratory
- RNS: Decrement
- SF-EMG: Increased jitter
- AChRα1L251R protein
- Expression: Reduced on muscle surface
- Channel properties: Conversion of ion selectivity from cationic to anionic
- AChR changes from excitatory to inhibitory channel
- Muscle biopsy: Type 2 fiber smallness
Abnormal ACh-AChR Interaction: Low-affinity, Fast-channel syndrome
● Acetylcholine receptor ε subunit (CHRNE)
● Acetylcholine receptor α subunit (CHRNA1)
- Genetics: AChR mutations
- ε AChR subunit
- α AChR subunit: Val132Leu; 381delC
- ε AChR subunit
- Physiology
- Reduced affinity of AChR for ACh: In open channel and desensitized states
- Affinity is mainly reduced in open & desensitized states of AChR
- Decreased rate, number & time of AChR channel openings
- Infrequent (Reduced) AChR openings during ACh occupancy
- Effects of mutation on channel function
- Brief AChR activation
- Resistance to desensitization
- Clinical features
- General: More severe than when main effect of mutation is on AChR gating
- Onset: Congenital; Fetal movements reduced
- Weakness: Generalized; Proximal > Distal; Face & Neck
- Episodic: Commonly develop life threatening episodes of apnea in childhood
- Ophthalmoplegia: Common
- Treatment
- 3,4-Diaminopyridine
- ? AChE inhibitors (Mestinon)
- Laboratory
- RNS:Decrement
- Postsynaptic defect in AChR: Numbers normal or Reduced
- NMJ morphology: Normal
CMS1B: Abnormal ACh-AChR Interaction; Fast-channel syndrome due to Gating abnormality
● AChR, α-subunit (CHRNA1)
- Mutations: Compound Heterozygous
- Neuromuscular junction
- Reduced Number of AChRs at NMJ
- Some endplate regions have sparse or shallow junctional folds
- Miniature endplate currents: Brief channel activation episodes
- Channel properties
- Slow opening
- Fast closure
- Reduced affinity for ACh: 2.5x
- M3 domain of AChR subunits: Regulates rates of channel opening & closing in volume-dependent manner
- Clinical
- General: Milder than when AChR affinity for ACh is low
- Onset: Birth; Extraocular muscle paresis
- Fatigability: Since early childhood
- Weakness: Face; Neck flexor; Trunk; Limbs
- Treatment: 3,4 Diaminopyridine + Mestinon
CMS1B: Fast-channel MG syndrome with Decreased probability of AChR channel opening
● AChR, α-subunit (CHRNA1)
- Mutations: Heterozygous α subunit
- αV132L (Cys-loop (β-hairpin) domain)
- α381delC: Low expressor; Null mutation
- Neuromuscular junction
- Normal number of AChRs at NMJ
- Endplate ultrastructure normal
- Miniature endplate currents
- Brief channel activation episodes
- Small amplitude
- AChR properties
- Opening probability after interaction with ACh reduced
- Low affinity for ACh
- Increased resistance to desensitization
- Cys-loop of AChR subunits: Highly conserved domain
- Clinical
- Onset: Birth
- Fatigability: Since early childhood
- Weakness: Diffuse; Respirator dependent
- Repetitive nerve stimulation: 25% to 50% decrement
- Treatment: Partial response to Mestinon
High Conductance & Fast Closure of AChRs: Congenital
- Weakness: Ocular, Neck and patchy Limb
- Postsynaptic defect in AChR channel kinetics
- Decrement in hand muscles post-exercise
- Little response to AChE inhibitors
CMS2B: Congenital MG, Beta subunit mutation; Fast Channel syndrome + Reduced AChR expression 52
● AChR, β-subunit (CHRNB1)
- Epidemiology: 1 patient
- Genetics
- Inheritance: Recessive
- Mutations
- c.812C>T (p.P248L ((p.P271L)): In M1-M2 linker
- 430 bp frame shift deletion (Loss of exon 8)
- CHRNB1
- Functions: Channel gating
- βP248L mutation
- AChR channel opening time: Reduced to 36% of normal
- MEPP decay time: 22% of normal
- M1-M2 linker: Contributes to elementary state transitions that open & close AChR channel
- Clinical
- Onset age: Congenital
- Weakness
- Severe
- Respiratory
- Bulbar: Dysphagia; Speech hypophonic
- Gait: Onset 5 years; Ambulatory
- Fatigability
- Muscle size: Small
- Joint contracture: Ankles
- Laboratory
- RNS (2 Hz): 10% to 37% decrement in different muscles
CMS2C: Congenital MG, Beta subunit mutations; Reduced expression of AChRs & Severe weakness
● AChR, β-subunit (CHRNB1)
- Epidemiology: 1 family, 3 patients
- Genetics
- AChR Mutations: Compound heterozygosity
- β1276DelEQE
- Located in long cytoplasmic loop between transmembrane domains M3 & M4
- Disrupts interaction between β &δ subunits
- Impairs AChR assembly
- Reduces incorporation of AChRs into surface membrane: More AChRs remain in cytoplasm
- Exon 8 deletion: Abolishes expression of pentameric AChR
- β1276DelEQE
- Allelic disorders
- AChR Mutations: Compound heterozygosity
- Clinical
- Onset: Birth
- Weakness: Severe; Respiratory failure; Poor feeding
- Repetitive nerve stimulation: Decrement
- Treatment: Partial response to acetylcholinesterase (AChE) inhibitors
- Course
- Static weakness
- Survival at least through childhood with support of vital functions
- Neuromuscular junction pathology
- More small endplate regions
- Endplate regions distributed over 3x increase in muscle fiber surface
- NMJ ultrastructure
- Structure: Normal presynaptic & post synaptic
- Size: Small presynaptic terminals; Small postsynaptic area
- AChR #: Reduced to 7% of normal
- NMJ physiology
- AChR channels: 89% normal; 11% with features of fetal AChRs (containing γ subunits)
- Quantal release by impulse: Normal
- Miniature endplate potentials (MEPPs): Reduced amplitude
- AChR opening time: Normal MEPCs
CMS3B: Congenital MG, Delta subunit mutations; Fast-channel MG syndrome + Arthrogryposis multiplex congenita
● AChR, δ-subunit (CHRND1)
- Mutations: Heterozygous δ subunit
- δ756ins2: Null mutation
- δE59K
- Short channel activations: Predict fast decay of endplate currents
- Dysfunction of fetal & adult AChRs
- Clinical features
- Reduced fetal movements
- Birth: Joint contractures, Hand IP joints
- Weakness
- Onset: Congenital
- Cranial nerve: Poor feeding; Ophthalmoplegia; Dysphagia
- Respiratory failure
- Distribution: Arms & Legs; Proximal & Distal
- Fatigue
- Laboratory
- Tensilon test: Positive
- SFEMG: Abnormal, increased jitter
- Treatment: Anti-AChE medications
- Other Arthrogryposis syndromes with familial MG
- Recurrent congenital MG: Maternal antibodies vs fetal AChRs
- Multiple pterygium syndrome (Escobar): AChR γ-subunit mutations
- Rapsyn mutations
CMS3B: Congenital MG, Delta subunit mutations; Reduced expression of AChRs & Fast channel effects
● AChR, δ-subunit (CHRND1)
- Epidemiology: Saudi Arabian patients
- Genetics
- Mutation: P250Q missense in transmembrane domain 1; Homozygous
- Clinical
- Onset
- Severe weakness at birth
- Respiratory insufficiency or failure
- Hypotonia
- General: Small habitus; Large ears; Micrognathia; High-arched palate
- Weakness
- Ophthalmoplegia: Nearly complete
- Ptosis: Moderate
- Facial diplegia
- Bulbar: Markedly weak lingual & masticatory muscles
- Neck flexion: Very weak
- Limb muscles: Moderate weakness
- Improvement with age
- Onset
- Laboratory
- Channel disorders
- Reduced numbers of AChRs at NMJs
- 16% to 34% of normal
- Impaired δ/α subunit association during early AChR assembly
- Fast channel effects: Changes in gating & ACh binding
- Hindered opening of the doubly occupied closed receptor (A2R)
- Rapid dissociation of acetylcholine from A2R
- Fast decaying, low-amplitude endplate currents
- Abnormally brief channel opening events
- NOTE: Similar mutations in ε-AChR subunit have slow channel effects
- Presynaptic: Number of quanta released by nerve impulse
- Low-normal range
- Differs from high normal range in α, β & ε mutations
- Reduced numbers of AChRs at NMJs
- Pathology
- Multiple large endplate regions on each muscle fiber
- Some reduction of 2° postsynaptic folds
- Type II muscle fiber atrophy: Mild
- Repetitive nerve stimulation: Decrement
- Treatment: Partial improvement with combined pyridostigmine & 3,4-diaminopyridine
- Channel disorders
- Other: AChR Delta subunit mutation syndromes
CMS8: Congenital MG, Agrin mutations
● Agrin (AGRN)
- Nosology: Myasthenic syndrome, congenital, with pre- & postsynaptic defects (CMSPPD)
- Epidemiology: ≥ 10 families; 0.8% of Congenital Myasthenic syndromes
- Genetics
- Mutations
- Missense common in at least one allele
- Glu42Ala, Ala1506Thr, Gly1709Arg, G76S, Val1727Phe & Nonsense; Often homozygous
- Allelic disorder
- Fetal akinesia: Null mutations
- Mutations
- Agrin protein
- Heparin sulfate proteoglycan
- Mutant protein: Destabilized NMJs; Normal binding to α-dystroglycan
- Clinical
- Onset
- Age: Early childhood
- Difficulty running
- Weakness
- Severity: Mild
- Distribution
- Limbs
- Proximal & Distal
- Some patients predominantly distal: Arms or Legs
- Symmetric
- Limbs
- Face (50%)
- Axial: Uncommon
- Respiratory: Some patients; Moderate
- Muscle atrophy: Distal predominant
- Fatigue
- Eyes: Ptosis; Normal EOM & Pupils
- Exacerbations: Menstrual periods; Pregnancy
- Overall course: Non-progressive
- Onset
- Milder patients: No scoliosis or contractures
- Intellectual disability: Few patients
- Treatment
- Salbutamol (2 - 3 mg tid)
- Ephedrine (50 md/day x 3 days, then 2 mg/kg qAM)
- Anti AChE drugs: No benefit; Worsening reported
- 3,4-diaminopyridine: Small benefit
- RNS
- 3 Hz: Decrement (Magnitude 10% to 72%)
- 30Hz: No potentiation or Increment in 1 patient
- Post-exercise increment: Up to 285%
- EMG: Early recruitment
- NCV: Normal; Single stimuli produce single, normal amplitude CMAPs
- Muscle pathology: Disorganized NMJs
- Nerve-terminals
- Diameter decrease
- Neurofilament destructuring
- Sprouting
- Postsynaptic
- Terminal Schwann cells: Cytoplasmic extensions or Absent
- Synaptic gutters: Fragmented
- Synaptic remodeling
- Muscle fibers
- Varied fiber size
- Myotendinous junctions: Large, Irregular
- Type 1 fiber predomiance
- Nerve-terminals
- MRI: Gastrocnemius & Semitendinosis atrophy
- Epidemiology: 2 patient
- Genetics
- Inheritance: Recessive
- Mutations: Nonsense (Null)
- Clinical
- Still born fetus
- Laboratory
- Prenatal ultrasound: Hypoechoic muscle; Joint contractures
CMS9: Congenital MG, MuSK mutations; Reduced expression of AChRs
● MuSK
- Epidemiology: Two families
- Genetics
- MuSK mutations
- Frameshift mutation: c.220insC
- Missense: V790M; P344R
- Allelic disorder: Fetal Akinesia Deformation Sequence 1 (FADS1)
(Arthrogryposis)
- MuSK mutations
- MuSK protein functions
- Tyrosine kinase receptor
- Role in aggregation of molecules in postsynaptic NMJ membrane
- AChRs
- ErbB family receptor tyrosine kinases
- Presynaptic differentiation: Absence leads to aberrant innervation
- Mutated MuSK protein: Increased degradation rate
- Clinical
- Onset: Neonatal
- Motor
- Respiratory involvement: Episodes during 1st year
- Bulbar involvement: During childhood; Episodic
- Ocular
- Ptosis
- Eye movement limitation (100%): Variable Up & Lateral gaze
- Weakness
- Face: Mild
- Arms > Legs
- Proximal > Distal
- Neck extensors
- Respiratory: Neonatal & Later in course
- Fatigue
- Course
- Improvement during childhood
- Exacerbation during pregnancy
- Progressive in adulthood
- Treatment
- 3,4-diaminopyridine: 50 mg per day
- No benefit: Acetylcholinesterase inhibitors
- Laboratory
- Pathology
- Nerve terminals
- Aberrant branching
- No 200 kD neurofilament subunits
- Several AChR patches on surface of single fibers
- Nerve terminals
- Molecular
- Acetylcholine receptors
- ε-subunit: Reduced expression at NMJs
- γ-subunit: Upregulated
- MuSK: Reduced at NMJs; Increased in perijunctional regions
- Acetylcholine receptors
- Family history: Sibling with congenital MG
- Neonatal hypotonia
- Respiratory failure
- Vocal cord paralysis
- Death 1.5 years
- Other: Myasthenia gravis with serum IgG binding to MuSK
- MuSK DNA polymorphisms: Patient with MuSK-seropositive autoimmune myasthenia gravis
CMS11: Congenital MG, Rapsyn mutations; Reduced expression of AChRs (CMS1E)
● Rapsyn
- Rapsyn genetics
- ~45 mutations identified
- Mutation type: Missense & Insertion
- Mutation actions
- Diminished co-clustering of AChR with rapsyn
- Normal rapsyn self-association
- Tetratricopeptide repeats (TPRs) of rapsyn: Subserve self association; L14P, N88K, 553ins5
- N88K: Present in many patients of Western European ethnic origin
- E-box of RAPSN promoter
- Mechanism: Regulate transcriptional activities
- -38A to G mutation: Homozygosity in Near-Eastern Jews
- Marked facial malformations
- Mild course
- -27C to G mutation
- Other: R164C; L283P
- Allelic disorders
- FADS2 (Arthrogryposis; Fetal Akinesia Deformation Sequence)

- Congenital MG: Reduced AChRs at NMJs
- Congenital MG with Facial Malformations
- Congenital Limb-Girdle MG with Digenic RAPSN & AK9 mutations
- FADS2 (Arthrogryposis; Fetal Akinesia Deformation Sequence)
- 43-kDa postsynaptic protein
- Function: Clustering of AChRs at the NMJ
- Structure
- Tetratricopeptide repeats (TPRs): Subserve self association; 7 repeats
- Coiled-coil domain: Binds to AChR
- RING-H2 domain: Associates with β-dystroglycan; Links rapsyn to subsynaptic cytoskeleton
- Onset
- Age: Birth or Infancy
- Weakness: Ptosis; Respiratory; Bulbar
- Severity
- Mild to Severe
- May vary with same mutation
- MG at birth: Early onset group
- May have same mutations as later onset group
- Prenatal: Reduced fetal movements
- Motor
- Hypotonia
- Weak suck & Cry
- Delayed motor milestones
- Ptosis
- Joint & Skeletal
- Arthrogryposis: Most patients
- Palate: High arched
- Spine: Scoliosis or Lordosis
- Episodic crises
- Often with respiratory failure
- Precipitated by: Infection, fever or no obvious event
- Less ophthalmoplegia than AChR ε-subunit mutations
- Early: Respiratory failure
- Improvement with age
- Walk in 2nd year
- Reduced contractures
- Less severe long term disability than AChR ε-subunit mutations with early onset
- Onset: 13 to 48 years
- Eyes: Ptosis without ophthalmoparesis
- Weakness
- Mild to moderate
- Diffuse or Proximal
- Masticatory muscles
- Face strength may be normal
- Normal early motor milestones
- Fatigue
- Tendon reflexes: Present
- Mouth (-38A to G mutations): Mandibular prognathism; Malocclusion; High-arched palate; Crowded teeth
- Differential diagnosis: Sero-negative immune myasthenia gravis
- Often benign with anti-AChE treatment
- Occasional exacerbations: Temporary
- Rare patient: Death in Childhood
- Anti-AChE medications: Incomplete benefit
- 3,4 Diaminopyridine: May add to benefit from anti-AChE treatment
- Pathology
- Type 1 muscle fiber preponderance
- Decreased staining at NMJs for
- Rapsyn
- AChRs: 8% to 48% of control; Less severe reduction than with AChR mutations
- Normal AChE
- Postsynaptic morphology
- NMJ extended over increased length of muscle fiber
- Increased number of end plate regions
- Simplified post synaptic membrane: Reduced postsynaptic area & secondary clefts
- Repetitive nerve stimulation
- Decrement
- 50% to 80% of patients
- Normal: More frequent in
- Congenital onset patients
- Muscles with normal strength
- Abnormal: Most later onset patients
- Other
- Decrement after exercise or subtetanic potentiation (10 Hz)
- SFEMG abnormality: Most patients
- Decrement
- Post synaptic physiology

From M. Sadeh
Congenital MG with
Facial Malformations- MEPPs: Small
- AChR function: Normal
- ACh release: Normal
- Zebrafish, Danio rerio
- Mouse rapsyn knockout
- Absence of initiation of postsynaptic differentiation
CMS: Congenital MG with facial malformations
● Rapsyn
- Nosology: See Congenital MG with rapsyn mutations
- Genetics: -38A to G mutations
- Epidemiology: Inbred Iraqi & Iranian Jews
- Clinical
- Extraocular
- Ptosis
- No ophthalmoplegia
- Cranial nerves
- Facial weakness
- Dysarthria
- Weak mastication
- Face
- Mandibular prognathism
- Palate: High arched
- Face: Elongated
- Treatment: AChE inhibitors
- Course: Stable & Benign
- Extraocular
- Electrophysiology
- Repetitive nerve stimulation
- Decrement: Some patients
- Single fiber EMG: Abnormal jitter
- Repetitive nerve stimulation
CMS: Congenital Limb-Girdle MG with Digenic RAPSN & AK9 mutations 47
● Rapsyn (RAPSN)
● Adenylate kinase 9 (AK9)
- Epidemiology: 1 family, 2 patients
- Genetics: Mutations
- RAPSN: Arg164His, Homozygous
- AK9: c.332-14A>G, Homozygous, Start-gain mutation; Introduces cryptic 5'-UTR signal in intron 5
- AK9 protein
- Cytosolic
- Catalyzes phosphorylation of AMP, dAMP, CMP & dCMP with ATP as phosphate donor
- Mutation: May reduce nucleotide sugars for N-glycosylation pathway
- Clinical
- Onset age: 2nd decade
- Motor
- Weakness: Proximal; Arms > Legs; Symmetric
- Walking difficulty
- Fatigability
- Falls: Occasional
- Atrophy: Proximal muscles
- Eyes: Normal
- Treatment: Pyridostigmine; Salbutamol
- Course: Slow progression or Stable
- Laboratory
- Tensilon test: Positive
- Muscle biopsy: "Unremarkable"
- Chest CT: Thymus hyperplasia
CMS15: Familial Myasthenic Syndrome (CMSWTA)
● Alg14 UDP-N-Acetylglucosaminyltransferase subunit (ALG14)
- Epidemiology: 4 families
- Genetics
- Mutations: Pro65Leu & Arg104X; Asp74Asn, Val141Gly, Arg109Gln
- Allelic disorders
- CMS15
- Intellectual developmental disorder + Epilepsy, Behavioral abnormalities & Coarse facies (IDDEBF)
- Myopathy, Epilepsy & Progressive Cerebral Atrophy (MEPCA)
- ALG14 protein
- Interacts with DPAGT1
- ALG7/13/14 complex: Multiglycosyltransferase
- Catalyses 1st two steps in biosynthesis of LLO precursor for N-glycan assembly
- Asparagine (N)-glycosylation
- Enriched at NMJs: Localized with α-Bungarotoxin
- Mutation: Causes ~50% reduced numbers of AChRs at NMJs
- Clinical
- Onset age
- CMG syndrome: 1st decade
- CMG + CNS: Antenatal or Infancy
- Muscle & Strength
- Early: Hypotonia
- Limbs: Moderate weakness, Proximal & Distal
- Difficulty climbing stairs & running
- Face & Bulbar: Strong
- Eye: Divergent exophoria; No ophthalmoplegia; ? Ptosis
- Severe involvement: Respiratory failure
- Contractures: Congenital; Knees & Elbows
- CNS: Some patients
- Epilepsy: Therapy resistant
- Course
- CMG: Stable or Mildly progressive
- CMG + CNS: Death < 1 year
- Treatment
- Anti-AChE medication (Pyridostigmine): Benefit may be transient
- Onset age
- Laboratory
- Serum CK: Normal
- RNS: 19% Decrement
- SFEMG: Jitter & Blocking
- Muscle biopsy
- Type 2 fiber predominance
- Atrophy
- Tubular aggregates: Some patients 172
- CNS
- Cerebral atrophy: Progressive
- Delayed myelination
- Astrocytosis
- Neuron loss: Multifocal
- EEG
- Burst-suppression
- Hypsarrhythmia
- ALG14 variant: Myopathy, Epilepsy & Progressive Cerebral Atrophy (MEPCA)
- Epidemiology: 6 infants, 4 families
- Genetics
- Inheritance: Recessive
- Mutations: Missense; Asp74Asn (Common), Val141Gly, Arg109Gln, Ile175Asn
- Clinical
- Onset: Birth, in utero, or Postnatal
- Pregnancy: Polyhydramnios; Reduced fetal movements
- Hypotonia
- Respiratory insufficiency
- Feeding problems
- Ptosis (50%)
- Skeletal
- Joint contractures; Foot deformities
- High-arched palate
- Dysmorphism: Turricephalus, Scaphocephalus, Micrognathia
- Seizures: Refractory
- Pyridostigmine treatment: Transient improvement
- Death: < 1 year in most
- Laboratory
- EEG: Burst suppression; Hypsarrhythmia
- Brain imaging: Supratentorial atrophy, progressive; Myelination delay
- EMG: Denervation
- RNS: Decrement
- Muscle pathology
- Type II fibers: Predominance
- Fiber sizes: Atrophy & Varied
- Internal architecture: NADH punctate
- Mitochondria: Normal
- Brain pathology: Reactive astrocytosis, Microglia, Decreased myelination
CMS16: Weakness + Episodic apnea & Bulbar dysfunction
● Sodium Channel - α subunit (SCN4A)
- Epidemiology
- 3 families
- Genetics
- SCN4A gene mutations
- Missense: V1442E; R1454W; R1457H; R1460Q; R1460W
- Loss of function
- Associated mutation: Probable polymorphism: S246L
- SCN4A, α-subunit: Allelic disorders
- SCN4A gene mutations
- Clinical
- Onset
- Age: Infancy
- Hypotonia
- Episodic respiratory failure
- Episodic attacks
- Sudden onset
- Length: 3 to 30 mintues
- Weakness
- Respiratory failure
- Bulbar dysfunction
- Weakness
- General
- Feeding
- Face; Trunk; Limb
- Proximal & Distal
- Ocular: Ptosis: Limited eye movements
- General
- Fatigue: Rapid over minutes
- Myalgia
- Skeletal
- High-arched palate
- Adduction deformity: Knees; ankles
- Lumbar lordosis: Increased
- CNS
- Mental retardation: ? Related to apneic episodes
- Onset
- SCNA4: Micro-electrophysiology
- AChRs at NMJs: Normal number & function
- Muscle fiber membrane potential: Normal
- MEPP amplitude and quantal content of the EPP: Normal
- Failure of action potential stimulation by normal EPP
- Depolarization of membrane to -40mV: Fails to trigger action potentials
- Na channels
- Normal density
- Trapping of mutant channels in fast-inactivated state
- Laboratory
- Normokalemia
- CMAP decrement: Normal to 85%
- 10-Hz stimulation for 5 min
- 50-Hz stimulation for 2 sec
- 2-Hz stimulation
- No decrement in rested muscle
- 50% decrement after 10-Hz conditioning stimuli for 1 min
- SF-EMG: Abnormal jitter
- Muscle pathology
- Type 1 muscle fiber smallness
- NMJs: Normal presynaptic terminals & Postsynaptic folds
- Vacuoles: Non-rimmed
- MRI: Mild cerebral atrophy
- Carriers of recessive mutation: Myotonia on EMG
CMS17: Congenital MG, LRP4 mutations
● Low density lipoprotein receptor-related protein 4 (LRP4)
- Epidemiology: 2 families; 3 patients
- Genetics
- Mutations
- Missense: Glu1233Lys, Arg1277His
- Location: 3rd LRP4 β-propeller domain
- Mutations
- Allelic with
- Cenani-Lenz syndactyly syndrome
- Sclerosteosis 2
- Cenani-Lenz syndactyly syndrome
- Mutation effect: Impairs binding of LRP4 to agrin & MuSK
- Antibody-related disorder: AChR Antibody-negative MG
- Onset
- Age: Congenital
- Respiratory & feeding insufficiency
- Fatigue: Walking distance limited
- Weakness: Moderate; Proximal > Distal; Respiratory
- Eyes
- Ptosis
- EOM: Limitation of lateral movements, mild
- Treatment
- Albuterol: Improved strength
- Pyridostigmine: Causes increased weakness
- Course: Intermittent use of wheelchair as young adult
- Repetitive nerve stimulation: Decrement in proximal muscles
- Muscle
- Histology: Type I muscle fiber predominance
- AChRs at NMJs: Normal distribution; Numbers slightly reduced; Channel function normal
- NMJ ultrastructure: Reduced size of presynaptic nerve terminal & postsynaptic region
CMS22: Familial Myasthenic Syndrome
● Prolyl endopeptidase-like (PREPL)
- Epidemiology: 22 patients; Female 66%
- Genetics
- Mutations
- Truncating > Missense, Large deletion
- c.1528c>T: 3 patients
- Allelic wtih: Recessive contiguous gene deletion syndromes
- Hypotonia-Cystinuria syndrome (HCS)
: with SLC3A1 disorder
- Atypical hypotonia–cystinuria syndrome: CAMKMT
- 2p21 deletion syndrome: PPM1B
; Most severe phenotype
- Hypotonia-Cystinuria syndrome (HCS)
- Mutations
- PREPL protein
- Oligopeptidase
- Interacts with: Clathrin-associated adaptor protein 1 (AP-1)
- Effector
- Binds to m1A subunit to release AP-1 from membrane target
- Mutations: PREPL protein absent from NMJs & Muscle fibers
- Clinical
- Onset age: Birth
- Hypotonia
- Feeding difficulties: Infancy
- Face: Eyelid ptosis; Tented upper lip
- Weakness
- Proximal & Neck predominant
- Variable during day
- Gait: Waddling
- Dysarthria
- Tendon reflexes: Normal
- Growth: Slowed
- Cognitive impairment: 50%
- Treatment
- AChE inhibitors: Improved strength in younger patients
- Laboratory
- Repetitive nerve stimulation (2 Hz): No decrement in most
- End plate physiology: Pre- & Post-synaptic features
- Quantal content of endplate potential: Decreased
- Miniature endplate potentials: Reduced amplitude
- NMJ AChRs: Normal number
- EMG: Normal
- Endplate geometry: Normal
- Serum insulin-like growth factor (IGF)-1 level: Low
- Growth hormone: Deficiency
CMS: Familial Myasthenic Syndrome 51
● Chromodomain helicase DNA-binding protein 8/Duplin (CHD8)
- Epidemiology: 1 monozygous twin pair
- Genetics
- Mutation: Missense; c.1732C>T (p.R578C); Heterozygous; Duplin domain
- Inheritance: de novo
- Allelic disorders
- Intellectual dysability, Autism susceptibility, Overgrowth syndromes ± Macrocephaly
: Microdeletions
- Gastrointestinal disorders
- Intellectual dysability, Autism susceptibility, Overgrowth syndromes ± Macrocephaly
- CHD8 protein
- ATP-dependent chromatin remodelling enzyme
- Regulation of DNA accessibility & gene expression
- Transcriptional regulation of canonical WNT-pathway
- Inhibits binding of β-catenin to TCF4
- Cell distribution: Cytoplasmic, especially Golgi, NMJ & Subsarcolemmal
- Muscle
- Canonical WNT-signalling reduces rapsyn expression
- NMJ development
- Clinical
- Pregnancy: Pre-eclampsia
- Birth: Respiratory distress; Ptosis; Weakness
- Skeletal
- Head: Macrocephaly; Hypertelorism;
- Scoliosis
- GI: Constipation
- Motor
- Leg weakness: Moderate
- Walking: Onset 2.5 years; Waddling gait
- Deterioration: Onset 6 years
- Voice: Weak
- Fatigability: Improvement with Tensilon; Worse with cold
- EOM: Full
- Intelligence: Normal
- Drug treatment: 3,4-diaminopyridine (3,4-DAP; 0.5 mg/kg BW/d)
- Laboratory
- NCV: Normal
- RNS: No decrement
- Brain MRI: Normal
CMYO12: Lethal Congenital Myopathy
● Contactin-1 (CNTN1)
- Nosology: Compton-North congenital myopathy; MYPCN; CMYP12; CMYO12
- Epidemiology: 2 families
- Genetics: Mutations
- Frameshift
- Homozygous
- Mutations; c.871dupT; Intragenic deletion of exons 2 to 15 & 18 to 19
- Contactin-1 protein
- Glycosyl phosphatidylinositol (GPI)-anchored: PIGG-dependent
- Neural adhesion molecule
- Immunoglobulin superfamily
- Ligand: NOTCH1
- Muscle: Localized to NMJs in normals; Sarcolemmal membrane in dystroglycanopathies
- Nerve: Paranode
- Function: Role in neurite growth & establishment and stabilization of synaptic connections
- Contactin-1 autoantibody (IgG): Immune demyelinating polyneuropathy
- Clinical
- Pregnancy: Polyhydramnios; Growth retardation
- Birth age: 28 to 35 weeks
- Motor: No spontaneous movements or Intubation needed
- Muscle mass: Reduced
- Skeletal
- Multiple contractures (Arthrogryposis)
- Simian creases
- Arachnodactyly with overlapping fingers
- Camptodactyly
- Death: Days to Months
- Laboratory
- CK: Normal
- Muscle pathology
- Fiber size: Mild variation
- Fiber architecture: Internal clear regions
- Endomysial connective tissue: Normal or Mildly increased
- Staining: Absent Integrin α7, β2-syntrophin, α-dystrobrevin
Familial Limb-Girdle MG syndromes
- CMS10: Familial Limb-Girdle Myasthenia
11
● Dok-7 (C4ORF25);
Chromosome 4p16.3; Recessive
25
- Epidemiology: Dok-7 MG
- Common in different ethnic origins
- > 70 patients described
- Genetics: Dok-7 mutations
- Type: Usually frameshift; Occasional missense
- Location: Common in exon 7 (Frameshift c.1124_1127dupTGCC); Also exons 2, 3, 4, 5
- Some mutations are complex: Identifiable only in cloned complementary DNA
- Allelic disorder: Fetal Akinesia Deformation Sequence 3 (FADDS3)
(Arthrogryposis;
Multiple Pterygium syndromes)
- Dok-7 protein
- Location: Post-synaptic, cytoplasmic
- Structure: Functional domains
- N-terminal pleckstrin homology (PH): Membrane association
- Central phosphotyrosine-binding (PTB): Dok-7 induced activation of MuSK
- Large C-terminal domain: Contains multiple tyrosine residues
- Functions
- Required for neuromuscular synaptogenesis & maintenance of NMJs
- Induces: Activation of MuSK & Clustering of AChRs
- AChR β-subunit phosphorylation
- Clinical features
- Onset
- Age
- Common: Birth to 2nd year
- Range: Birth to 3rd decade
- Occasional: Decreased fetal movements
- Hypotonia; Falling; Gait disorder; Weakness
- May be acute or chronic
- Associated with: Pregnancy
- Age
- Weakness
- Proximal
- Arms & Legs
- Trunk & Neck
- Symmetric
- Difficulty walking: Waddling gait
- Respiratory: Common with Dok-7 mutations
- Face: Weak; Ptosis, may be asymmetric
- Extra-ocular muscles: Normal
- Fatiguable: Worse after exercise & emotional events
- Severity: Variable
- Mild: Static weakness
- Severe: Progressive generalized
- Course
- Progressive: Especially in severe patients
- Intermittent exacerbations: Duration days to weeks
- Proximal
- Other muscle features
- Muscle wasting
- Discomfort: Cramps; Pain
- Skeletal
- Scoliosis
- No joint contractures
- Occasional systemic features
- Contractures
- Cardiac repolarization defect
- Treatments
- Ephedrine
- Salbutamol (Albuterol)
- Adults: 2mg tid or 4 mg bid (6-18 mg/d)
- Children (up to 6 years): 0.1-0.3 mg/kg/d
- Fluoxetine: 40 mg/d; Partial responses reported
- ? AChE inhibitors
- May benefit or worsen some patients with limb-girdle MG syndromes
- Dok-7 mutations
- No long-term benefit
- May have positive tensilon test
- Onset
- Laboratory
- Serum CK: Normal to Mildly high; 160 to 320
- Electrophysiology
- RNS: Decrement
- SF-EMG: Often increased jitter
- Muscle fiber irritability: Some patients
- Muscle pathology
- Internal nuclei
- Neuromuscular junctions
- Small size: Some
- Post-synaptic folds: Reduced
- Single or Multiple
- EM
- Destruction & remodeling of EPs
- Degeneration of post-synaptic folds
- Similar to: Slow channel syndromes
- Dok-7 protein
- Dok-7 mutations: Normal or reduced amounts at NMJs
- May be reduced with AChR mutations
- AChR function: Normal
- Presynaptic function: Normal
- Tubular aggregates: Not present with Dok-7 mutations
- Epidemiology: Dok-7 MG
- CMS12: Familial Limb-Girdle Myasthenia with Tubular Aggregates (CMSTA1)
32
● Glutamine:Fructose-6-phosphate Amidotransferase 1 (GFPT1);
Chromosome 2p13.3; Recessive
- Epidemiology: > 75 patients, 40 families
- Genetics
- Mutations: Missense, Splice site or Stop
- Location: Most regions of gene
- Loss of function mutations: More severe phenotype
- GFPT1 protein
- Hexosamine pathway: Rate-limiting enzyme
- Yields essential amino sugars for carbohydrate modifications
- End product of pathway: UDP-N-acetlyglucosamine (Glc-NAc)
- Substrate for N- & O-linked glycosylation
- Required for neuromuscular transmission
- Clinical
- Onset age
- Range: Infancy to 24 years
- 1st decade in 80%; Mean 8 years
- Normal at birth: Most patients
- Congenital onset: Some patients with severe weakness
- Weakness
- Proximal > Distal
- Shoulder & Pelvic girdle: 100%
- Distal weakness: 30% to 50%
- Forearm extensors, Intrinsic hand, Anterior leg
- Cranial
- Face: 30%
- Ptosis: 5%
- Ocular muscles: 0%
- Bulbar: 0%
- Neck: 25%
- Respiratory: 5% to 12%
- Permanent: > 30%
- Course
- Slowly progressive
- Prognosis
- Most (80%) remain ambulant
- Myopathic weakness with less response to CMS drugs
- Tendon reflexes: May be reduced
- Contractures: Occasional; Mild
- Sensation: Normal
- Treatments: Partial benefit
- Anti-AChE medications: More response than with Dok-7 mutations
- 3,4 Diaminopyridine
- Albuterol (6 mg/day)
- Onset age
- Laboratory
- EMG: Myopathic
- Motor units: Small amplitude; Short (Myopathic); Unstable
- Some patients: Spontaneous activity; Fibrillations & Positive sharp waves
- Repetitive nerve stimulation: Decrement
- SFEMG: Abnormal jitter & blocking
- Serum CK: Normal in most; Occasionally High
- Muscle MRI
- Fatty infiltration (T1w imaging)
- Nuscle involvement
- Non-selective
- Sparing: Adductor magnus; Semimembranosus; Gastrocnemius, medial
- May be near normal with severe weakness
- Muscle pathology
- Tubular aggregates (80%): Especially in Type 2 fibers, May be small
- Some patients
- Myopathic: Varied fiber sizes & Endomysial fibrosis
- Desmin or Dystrophin+ aggregates
- Rimmed vacuoles (30%)
- Ultrastructure: Autophagic & other vacuoles; Tubular aggregates; Granulofilamentous deposits
- Glycosylation & Glycoprotein expression
- Often reduced (70 kD band)
- Reduced O-glycosylation
- May be normal
- α-dystroglycan staining: Normal
- Sialylation of transmembrane proteins: Reduced in extra-junctional regions
- Increased sarcolemma staining for peanut agglutinin (PNA)
- NMJs
- AChRs: Reduced or Normal numbers
- Morphology
- Normal or Post-synaptic simplification (Loss of folds)
- Low nerve terminal area
- Post synaptic: Small; Single or grape-like regions
- Multiple AChE regions
- Neuropathic features: Fiber type grouping in 30%
- EMG: Myopathic
- CMS13: Familial Limb-Girdle Myasthenia with Tubular Aggregates 2 (CMSTA2)
34
● Dolichyl-phosphate N-acetylglucosamine phosphotransferase (DPAGT1);
Chromosome 11q23.3; Recessive
- Epidemiology: 11 families; 18 patients
- Genetics
- Mutations: Missense & Duplication
- Allelic with: Congenital disorder of glycosylation, type IJ
- DPAGT1 protein
- Expression: Ubiquitous
- Catalyzes first committed step of N-linked protein & lipid (LLO) glycosylation
- Phosphorylated UDP-GlcNAc (from cytoplasmic UDP-GlcNAc) is added to dolichol phosphate (a transmembrane lipid carrier)
- Dolichol-PP-glycan assembly
- Required for efficient
- Glycosylation of acetylcholine-receptor subunits
- Export of AChRs to cell surface
- Clinical
- Onset age: 0.5 to 17 years; Median 2 years; Normal at birth
- Early: Hypotonia
- Weakness
- Proximal: Most patients
- Distal: Many patients
- Arms ≥ Legs
- Gait disorder
- Neck flexion
- Respiratory: Moderate
- Face or Bulbar: Mild or None
- Course: Slowly progressive
- No: Ptosis or ophthalmoplegia;
- Skeletal
- Scoliosis: 50%
- Joint contractures: 40%
- Treatment: Cholinesterase inhibitors; 3,4-Diaminopyridine
- Course: Slow progression or Improvement
- Laboratory
- AChRs at NMJs: Reduced
- Repetitive nerve stimulation: Decrement (30% to 40%)
- SFEMG: Jitter & Blocking
- Repetitive discharges after single stimulation: 1 patient
- EMG: Myopathic
- Serum CK: Normal
- Transferrin IEF: Normal or Mildy reduced glycosylation
- Muscle pathology
- Tubular aggregates: More common in older patients
- ORAI1: Hypo-glycosylation; Over-activation
- NMJs
- Postsynaptic folding: Reduced
- AChRs: Reduced number
- AChE: Normal
- Glycogen: Increased
- Autophagic vacuoles
- CMS14: Familial Limb-Girdle Myasthenia with Tubular Aggregates 3 (CMSTA3)
37
● ALG2;
Chromosome 9q22.33; Recessive
- Epidemiology: 5 patients in 2 families
- Genetics
- Mutations: Val68Gly; c.214_226delinsAGTCCCCGGC, p.72_75delinsSPR; Homozygous
- Allelic with: Congenital disorder of glycosylation 1i (GDG 1i)
- ALG2 protein
- α-1,3-mannosyltransferase
- Catalyses early steps in asparagine-linked glycosylation
- Location: Enriched at NMJs in muscle
- Mutant: Reduced ALG2 protein in muscle
- Clinical
- Onset age: Infancy to 4 years
- Weakness
- Early: Delayed motor milestones; Hypotonia
- Pattern: Proximal > Distal
- Face: Mild
- Respiratory: Mild involvement
- Eye: Normal; Full EOM; No ptosis
- Tendon reflexes: May be reduced
- Skeletal
- Contractures: Knees (60%)
- Spine: Generally normal
- Joint Laxity: Distal (60%)
- Palate: High arched
- Symptoms exacerbated by: Stress; Infection
- Progression: Slow; 80% never ambulate
- Treatment: AChE inhibitors
- Laboratory
- Muscle
- Fiber size: Varied
- Myopathic
- Tubular aggregates (50%)
- Internal nuclei (50%)
- Type 1 predominance (50%)
- Repetitive nerve stimulation: Decrement up to 67%
- SFEMG: Jitter & Blocking
- Serum CK: Normal to Slightly high
- Muscle
Familial MG Syndromes: Other
- Familial Immune MG
60
- Epidemiology
- + Family history in 3% to 5.7% of MG
- May occur in same generation
- Sibs or Cousins; Parent/Child
- 2 people per family
- Clinical
- Onset age
- Mean: 12 years earlier than non-familial autoimmune MG
- Age range: 2 to 70 years
- Clinical features
- Similar to non-familial adult MG
- Generalized MG: 60% to 80%
- Severity: Worse than non-familial at disease onset
- Treatments: Usually responsive to Pyridostigmine & Corticosteroids
- Onset age
- Laboratory
- Serum Anti-AChR antibodies: 60%
- Thymus: Hyperplasia
- Rule out
- Variant syndrome: Familial Autoimmune Myasthenia Gravis
35
● Ecto-NOX Disulfide-Thiol exchanger 1 (ENOX1); Chromosome 13q14.11; Recessive
- Epidemiology: 1 Sicilian family
- Mutation
- c.*35A>G: Located in 3' UTR of ENOX1 gene
- Also present in 1 unaffected sibling
- Clinical
- Onset ages: 51 to 70 years
- Ptosis
- Eye movements: Diplopia; Fatigability of upward & lateral gaze
- Weakness: Neck; Extremities
- Fatigability
- Treatments: Pyridostigmine; Corticosteroids
- Laboratory
- AChR antibodies: 75% positive
- Repetitive nerve stimulation: Decrement
● Also see
- Epidemiology
- Reduction in number of AChRs at NMJs and Paucity of Secondary Synaptic Clefts
- Presynaptic defects in ACh synthesis, mobilization or storage
CMS: Reduced numbers of AChRs at Neuromuscular Junctions
● Acetylcholine receptor α subunit (CHRNA1)
● Acetylcholine receptor β subunit (CHRNB)
● Acetylcholine receptor δ subunit (CHRND)
● Acetylcholine receptor ε subunit (CHRNE)
● Receptor-associated protein of the synapse, 43-kD (RAPSN; Rapsyn)
- Most common type of congenital MG syndrome
- Genetics
- AChR mutations
- Often recessive
- Subunits: Usually ε mutations; Occasional α, β, or rarely δ, mutations
- May be compound heterozygous or homozygous
- Types
- Premature termination of translation: Frame shifting; Splice site; Stop codon
- Missense
- Signal peptide region (εG-8R)
- Amino acids needed for assembly of pentameric AChR: εS143L; εC128S; εR147L
- Affecting both AChR expression & kinetics
- Point mutations: Promoter region of subunit genes
- ε subunit nonsense mutations
- ε1101insT
- Reduced AChR expression
- Normal channel function
- ε1293insG
- No channel function
- c.1327delG: Common in Bulgarian Roma & Southeastern Europe 58
- Other frameshifting mutations
- Many patients are heteroallelic
- Compensatory mechanism: Expression of γ AChR (immature) at NMJs
- ε1101insT
- Rapsyn mutations
- AChR mutations
- Clinical features
- General: Varied, even with same mutation
- Severe form
- Onset: Birth
- Weakness
- Diffuse: Ocular; Bulbar; Respiratory; Limbs; Trunk
- Delayed motor milestones
- Respiratory insufficiency: Frequent
- Muscle bulk: Reduced
- Tendon reflexes: Normal or Reduced
- Skeletal
- Facial deformities: Prognathism; Malocclusion
- Scoliosis
- Intermediate form
- Onset: Early childhood
- Ocular paresis & Ptosis: Onset 1st year
- Weakness: Diffuse (Axial & Limbs); Moderate disability
- Bulbar: Dysphagia
- Fatigue
- Mild form
- Normal motor milestones
- Mild ptosis common
- Ophthalmoplegia: Partial
- Motor: Clumsy; Poor running
- Sensitivity to neuromuscular blocking agents
- Treatment
- AChE inhibitors
- 3,4-Diaminopyridine: 1 mg/kg/day in doses q 3 to 5 hours
- Less benefit in more severe disease
- Laboratory
- NMJ Morphology
- Endplate regions: Increased Number; Distributed over larger surface area of muscle fiber
- Most junctions: Integrity of post-synaptic membrane folds is preserved
- Occasional junctions: Endplate regions are simplified & small
- AChR on junctional folds: Patchy distribution; Reduced Density
- Physiology
- Reduced amplitiude of miniature endplate potentials & currents
- High quantal release by nerve impulse: frequent
- Fetal AChRs at NMJ containing γ instead of adult ε AChR subunits
- NMJ Morphology
CMS: Congenital MG syndrome resembling LEMS
- Epidemiology: 2 patients reported
- Genetics: Normal CACNA1A gene
- Clinical
- Onset: Birth
- Motor development: Delayed; Hypotonia
- Weakness: Bulbar & limb
- Mental retardation
- Electrophysiology: Repetitive nerve stimulation
- Small initial CMAP
- Rapid stimulation: Increment up to 5-fold
- Slow stimulation: Decrement
- Neuromuscular junctions
- AChRs: Normal number
- MEPPS: Normal amplitude
- EPP: Small quantal content; Increased after 40 Hz RNS
- 3,4 Diaminopyridine treatment
- Improves electrophysiologic defects
- Little clinical benefit
CMS7A: LEMS ± Nonprogressive Distal Motor Neuropathy
● Synaptotagmin 2 (SYT2)
- Epidemiology: 7 families
- Genetics
- Mutations: Ser306Leu; Asp307Ala; Pro308Leu; Ile371Lys; Inframe c.1082_1096del; Asp361_Leu365del
- Allelic disorder: Congenital onset presynaptic myasthenic syndrome, Recessive (CMS7B)
- SYT2 protein
- Calcium
- Binding
- Sensors in vesicular trafficking & exocytosis
- Secretory granules & Synaptic vesicles
- Binds: Botulinum toxins B & G
- Role in: Synaptic vesicle exocytosis
- Synchronous vesicle release at NMJs
- Mutant SYT2 protein
- Dominant-negative effect on synaptic vesicle exocytosis
- Disease expression may relate to disorder of presynaptic NMJ or Motor axon
- SYT1
mutations: Neurodevelopmental disorder (Baker-Gordon syndrome)
- Calcium
- Clinical
- Onset age: Childhood, early
- Foot deformities: Pes cavus; Hammer toes
- Weakness
- Proximal & Distal, or Distal only
- Legs > Arms
- May be asymmetric
- Gait disorder
- Ptosis: Few patients
- Motor: Other
- Fatigue: Improves with rest
- Atrophy: Leg
- Tendon reflexes: Decreased at ankles; Increase with exercise
- Sensory loss: 25%
- Course: Slow progression
- Treatment: 3,4-diaminopyridine variably effective
- Laboratory
- Electrodiagnostic
- CMAP amplitudes: Small; Increment after exercise
- SNAPs: Normal
- Repetitive Nerve stimulation
- SFEMG: Jitter increased
- EMG: Normal or Mild reinnervation
- Muscle
- Fiber sizes: Varied
- NMJs: Nerve terminals small & inclusions; Postsynaptic folding overdeveloped
- Electrodiagnostic
- SYT2 variant disorder: CMS7B, Congenital-onset Presynaptic myasthenic syndrome
53
- Epidemiology: > 10 patients
- Genetics
- Inheritance: Recessive
- Mutations: Loss of function; Biallelic
- Clinical
- General: May be more severe than Dominant syndromes
- Onset: Congenital
- Pregnancy: Reduced fetal movements
- Hypotonia
- Weakness: Severe
- Motor milestones: Delayed
- Face
- Axial
- Limbs: Distal or Proximal > Distal
- EOM: Slow; Limited
- General function: Minimal head control to Sitting
- Respiratory involvement: Common; Varied severity
- Tendon reflexes: Absent
- Sensation: Normal
- Skeletal: Scoliosis; Hip dislocation
- Treatment response: Varied/Mild
- AChE inhibitor (Pyridostigmine or Neostigmine)
- Salbutamol
- 3,4-DAP
- Cognition: Normal
- Laboratory
- Serum CK: Normal or Mildly high
- RNS: Decrement at 3 to 5 Hz; Increment with Short exercise
- NCV: CMAP amplitudes reduced
- EMG: Denervation or Myopathy
- Muscle pathology
- Fiber sizes: Varied; Angular fibers
- Tubular aggregates
- NMJs segmented
- Type predominance: 1 or 2
- Ultrastructure
- Convoluted postsynaptic membrane
- Membrane multivesicular bodies with densely packed synaptic vesicles & endosomes
- Brain MRI: Corpus callosum thin
CMS18 (DEE117): Myasthenia, Cortical hyperexcitability, Ataxia & Intellectual disability
● Synaptosomal-associated protein, 25-kD (SNAP25)
- Epidemiology: 1 patient
- Genetics
- Mutations
- Missense most common
- Ile67Asn: Produces SNAP25b splice variant
- Q117X: Likely produces truncated protein
- Probable mechanism: Dominant negative
- Allelic disorder
- Epilepsy & Encephalopathy, Dominant de novo
- Epilepsy & Encephalopathy, Dominant de novo
- Mutations
- SNAP25 protein
- Location: Nerve terminal; Presynaptic
- t-SNARE
- Attached to vesicle target plasma membrane
- Component of synaptic vesicle fusion machinery
- Loss of SNAP25
- Pool of vesicles primed for release: Empty
- Fast calcium-triggered exocytosis: Abolished
- Binds to: Munc13-1
- Splice variants
- SNAP25b: Plasma membrane of nerve terminals at motor endplates ¢ral synapses
- SNAP25a: Diffusely distributed in neuronal cytoplasm & axoplasm
- Mutation: MEPP frequency reduced
- Altered in: Schizophrenic brains
- Toxic cleavage: Botulinum toxins A & E
- Clinical
- Onset: in utero
- Hypomobility
- Polyhydramnios
- Birth: Cyanosis; Contractures; Arthrogryposis
- Motor
- Walking: Delayed; Limited
- Weakness: Fatigable
- Episodes: Ptosis, Blank stare, Unresponsive
- CNS
- Intellectual disability
- Ataxia: Dysarthria; Gait
- Spasticity
- Epilepsy
- Treatment
- Strength: Improved by 3,4-diaminopyridine, not pyridostigmine
- Seizures: Levetiracetam
- Course: Early death in patient with stop mutation
- Onset: in utero
- Laboratory
- EEG: Generalized atypical polyspike & wave discharges; Diffuse slowing
- Brain MRI: Normal
- Repetitive nerve stimulation: 18% to 50% decrement
- Muscle
- Histochemistry: Normal
- Synaptic areas: Pretzel-shaped or Ovoid; AChR & AChE normal
- NMJ ultrastructure: Normal
- Quantal release: Reduced probability
- Animals
- Blind-drunk (+/Bdr) mouse: Ile67Thr mutation
CMS19: Congenital Myasthenic Syndrome 19
● Collagen, type XIII, alpha-1 (COL13A1)
- Epidemiology: 16 families
- Genetics
- Mutations: Homozygous; Loss-of-function; c.1171delG, c.523-1delG
- COL13A1 protein
- Collagen
- α-chain of Atypical non-fibrillar collagen
- Expression: Low levels in many tissues
- Interacts with
- Extracellular matrix
- Proteins: Fibronectin, Heparin, Nidogen-2, Perlecan,15
- Anchored to plasma membrane: By a hydrophobic transmembrane segment
- Localized to NMJ: Postsynaptic membrane; Synaptic basement membrane
- Target of serum autoantibodies: Some patients with autoimmune MG
- Clinical
- Onset age: Birth
- Weakness
- Respiratory
- Crises: Fewer with increased age
- Vital cpacity: 14% to 62%
- Bulbar: Feeding difficulty; Face; Poor suck
- Truncal & Axial > Limbs
- Limbs: Arms & Legs; Proximal & Distal
- Milder patient: Distal arms
- Muscle bulk: Often reduced
- NO fluctuation or fatigability
- Course: Slow improvement ove years
- Respiratory
- Ocular
- Ptosis
- Ophthalmoplegia: Often mild; May be asymmetric
- Skeletal
- Dysmorphic: Micrognathia, Ears low-set, Palate high-arched; Face elongated & narrow
- Pectus carinatum & Pes cavus
- Spinal rigidity
- Scoliosis: Few patients
- Distal joint laxity: Few patients
- No contractures
- CNS: some patients
- Learning difficulties (50%)
- Motor milestones delayed (70%)
- Treatments
- 3,4-diaminopyridine
- Salbutamol
- Ventilatory support
- NOT: AChE inhibitors
- Laboratory
- Repetitive nerve stimulation: Decrement (70%)
- Jitter: Abnormal
- Serum CK: Normal
- Muscle biopsy
- Fiber size: Varied
- Immature muscle fibers
- NADH: Halo-like fibers
- Mitochondria: ? Large
- May be normal
- NMJ
- Presynaptic: Incomplete clustering of synaptic vesicles
- Postsynaptic: Defective maturation; AChE more extensive than AChRs
- Nerve terminals: Abnormal localization
- Brain & Muscle MRI: Normal
CMS24: Congenital Myasthenic Syndrome 24, Presynaptic
● Myosin IXA (MYO9A)
- Epidemiology: 3 patients, Kurdish & German families
- Genetics
- Mutations: Missense; .R1517H, R2283H, D1698G
- Allelic disorder: Arthrogryposis
- MYO9A protein
- Unconventional myosin
- Roles: NMJ development; Neurite extension & branching
- Location: NMJ, Pre- & Post-synaptic; Growth cones; Brain
- Single headed processive motor
- Tail region: Contains Rho-GTPase domain
- Postsynaptic: Binds to α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor (AMPAR) GluA2 subunit
- Clinical
- Onset age: Neonatal
- Hypotonia
- Apnea: Episodic
- Dysphagia
- Eyes: Variable
- Ptosis: Bilateral
- Ophthalmoplegia: Symmetric
- Nystagmus
- Oculomotor apraxia
- Weakness: Distal & Proximal
- Treatment: Partial responses to Pyridostigmine or 3.4-DAP
- Laboratory
- Repetitive nerve stimulation: Decrement in face
- SFEMG: Abnormal jitter
- EMG: Normal
- Brain MRI: Normal
- Serum CK: Normal
- MYO9A variant: Arthrogryposis
- Epidemiology: 1 family
- Mutations: c.6845G>A (p.Gly2282Glu); c.608A>G (p.Tyr203C)
- Clinical: Multiple limb contractures
CMS: Congenital Myasthenic Syndrome with Microcephaly & Cortical hyperexcitability
● Munc13-1 (UNC13A)
- Nosology: Neurodevelopmental disorder with Hhypotonia, Epilepsy & absent Speech (NEDHES)
- Epidemiology: 8 patients
- Genetics
- Mutation: Nonsense or Missense
- Allelic disorders
- ALS survival influence
- Neurodevelopmental disorder with Speech delay, Movement abnormalities & Seizures (NEDSMS), Dominant or de novo
- Munc13-1 protein
- Location: Presynaptic
- Active zones of NMJ nerve terminal & Glutamatergic synapses in brain
- Reqired for
- Synaptic vesicle release: Docking & Priming
- Brain development
- Binds to: Syntaxin 1B; SNAP25B
- Location: Presynaptic
- Clinical
- Onset
- Age: Birth; Premature
- Respiratory weakness
- Weakness
- Hypotonia
- Respiratory failure: Cyanotic episodes
- Feeding difficulties
- Ptosis
- Ophthalmoplegia
- Head
- Microcephaly: Circumference: < 5th percentile
- Forehead: High, Frontotemporal narrowing
- Nasal bridge: Wide
- Palate: High arched
- Jaw: Small
- Skeletal
- Thoracic kyphoscoliosis
- Clinodactyly: Fifth digits
- Contractures: Flexion; Proximal joints & knees
- CNS
- No speech
- Seizures
- Death: 50 months
- Onset
- Laboratory
- MRI: Corpus callosum thin
- EEG: Sharp waves
- Nerve conduction
- CMAP ampltudes: Small at rest
- RNS 2 Hz: 20% to 40% decrement
- RNS 50 Hz: 500% increment (0.8 to 4 mV)
- Microphysiology: NMJ
- MEPP frequency: 2% of normal
- Quantal content of EPP (m): 6% of normal
- α-bungarotoxin binding sites per EP: Normal
- Muscle
- Type 2 fibers: Small
- Type 1 fibers: Large
- Pre- & Post-synaptic morphology: Normal
CMS: Congenital Myasthenic Syndrome with Altered Synaptic Vesicle Homeostasis 49
● Rhabphilin 3A (RPH3A)
- Epidemiology: 1 patient
- Genetics
- Mutations: Missense; Arg269Gln, Val464Leu
- RPH3A protein
- Interacts with small GTP-binding proteins Rab3A & Rab27a
- Participates in trafficking & release of synaptic & secretory vesicles
- Clinical
- Onset age: Earl;y childhood
- Learning disabilities
- Cerebellar: Tremor, Gait ataxia, Dysmetria
- Bulbar
- Speech: Nasal
- Velopharyngeal insufficiency
- Obstructive sleep apnea
- Weakness; Mild; Neck, Proximal arms
- Fatigability: Responsive to albuterol sulfate
- Tendon reflexes: Normal
- EOM: Normal
- Abdominal pain
- Laboratory
- Hyperglycemia: Transient
- Repetitive nerve stimulation
- 2 Hz: Normal
- 30 Hz: 30% increase in CMAP area
- SFEMG: Jitter increased
- Muscle pathology
- Type 1 predominance
- Nonspecific filament bodies & mild Z-disk disruption in few fibers
- NMJ pathology
- Presynaptic terminals
- Synaptic vesicle density: Marked reduction in SV density,
- Double-membrane-bound sacs containing synaptic vesicles
- Abundant endosomes
- Degenerative lamellar bodies
- AChR & AChE: Normal
- Presynaptic terminals
CMS: Congenital MG with reduced quantal release by nerve terminals 16
- Onset: Infancy to 5 years
- Clinical
- Myasthenic symptoms
- Weakness
- Extraocular muscles: Normal
- Cerebellar: Trunk & limb ataxia; Nystagmus
- Myasthenic symptoms
- Electrophysiology
- Decremental response on 2-Hz stimulation: Not improved by stimulation at higher frequencies
- Normal first-evoked CMAP
- Marked decrease in number of quanta released by 1-Hz stimulation
- Treatment: Some patients improve with combined treatment with pyridostigmine & 3,4-DAP
CMS: Congenital MG with acquired MG developing in 4th decade 14
- Epidemiology +
- Single patient: HLA type DR3-B8-A1
- Sibling with congenital MG but no acquired disease: Thymectomy performed at 3 years
- Congential MG
- AChR mutations
- ε-subunit
- Heteroallelic
- Truncating: Y15X; 70insG
- Onset: Birth; Ptosis
- Slow motor development
- Weakness
- Severity: Moderate
- Distribution: Limb, Ocular & Bulbar
- Progression: None
- Laboratory
- Anti-AChR antibodies: None
- SF-EMG: Mild jitter
- Treatment: Anti-AChE agents (Pyridostigmine)
- AChR mutations
- Acquired MG
- Onset age: 34 years; After pregnancy
- Weakness
- Severe
- Increasing over 1 year
- Respiratory failure
- Laboratory
- Anti-AChR antibodies: Present; Bind to adult & fetal AChRs
- SF-EMG: Increased jitter
- NMJ pathology
- Reduced number of AChRs
- Multiple endplate regions on individual muscle fibers
- Treatment: Immunosuppression
Ocular Myopathy with Curare Sensitivity
- Epidemiology: 1 South Indian family, 9 patients
- Genetics: Recessive pattern of inheritance
- Clinical
- Onset age: Infancy or Early childhood
- Ophthalmoparesis
- Bilateral
- Often complete
- Ptosis
- Pupils: Normal
- Strength: Limbs
- Generally normal
- Weakness: Provoked by low doses of curare
- Proximal
- Dysphagia
- Ptosis: Increased
- 1 patient with fixed proximal weakness
- Tendon reflexes: Normal
- Some curare sensitive family members asymptomatic
- Laboratory
- EMG: Normal
- Repetitive nerve stimulation (Ulnar): No decrement
- Edrophonium test: Negative
- Muscle biopsy: Normal
MUTATIONS OF AChR SUBUNITS
|
Acetylcholine & Acetylcholine Receptors AChR subunit disorders: α, β, δ, ε, γ Diagrams Neuromuscular junction Neuromuscular junction disorders Neuromuscular junction molecules Also see: Myosin & related proteins |
α-subunit of AChR (CHRNA1): Mutations
● Chromosome 2q31.1
- Allelic disorders
β-subunit of (CHRNB1)
● Chromosome 17p13.1
- Genetics
- Mutations
- Often in exon 8
- Allelic disorders
- Slow channel syndromes: Dominant
- Fast channel syndrome: Recessive
- Congenital myasthenia gravis: Severe; Recessive
- Fetal akinesia deformation sequence (FADS): Recessive
- Myasthenia gravis, Acquired: Transcriptome association, rs4151121 56
- Mutations
ε-subunit of AChR (CHRNE)
● Chromosome 17p13.2
- Epidemiology
- ε subunit mutations: Most common cause of congenital MG syndromes
- Genetics
- Mutations
- Most patients compound heterozygotes
- Each patient with one null & one missense mutation
- North Africa: 1293insG founder mutation
- ε subunit: Only AChR subunit with null mutations in congenital MG
- Clinical: AChR dysfunction & phenotype correlates with missense mutation
- Allelic disorders
- Recessive: Congenital MG syndromes with
- AChR Deficiency (CMS4C)
- Fast channel syndromes (CMS4B)
- North Africa founder mutation
- Slow channel syndrome (CMS4A)
- AChR Deficiency (CMS4C)
- Dominant: Slow channel syndrome
- Congenital MG with acquired MG developing in 4th decade
- Recessive: Congenital MG syndromes with
- Mutations
- AChR function in disease syndromes: Changes with mutations
- Abnormal channel open times: Also see Slow channel syndromes
- Abnormal ACh-AChR interaction: Low-affinity, fast-channel syndrome
- Reduced AChR expression
- Postsynaptic reduction in number of AChRs at NMJs
- Short AChR open time
- Compensation for ε subunit loss: Allows some NMJ function & survival of patient
- γ subunit may be upregulated
- Mutations between M3 & M4 (ε1267delG):
Retention of intron 11 in some mRNA transcripts
- Restores reading frame
- Production of ε subunit at reduced level
- Partially rescues adult AChR function
- Clinical features
- Onset: Congenital
- Weakness: Variable severity
- Generalized: Proximal & Distal
- Face
- Ptosis
- Ophthalmoplegia
- Fatigue
- Laboratory
- Electrophysiology
- Decrement on RNS
- Slow channel syndrome also has repetitive CMAP response to single stimulus
- Pathology
- Post-synaptic folding: Normal
- Endplate morphology: Small, pale regions (Single or Multiple) distributed over larger region of muscle fiber
- Type 2 fiber atrophy
- Electrophysiology
- CHRNE variant syndrome: North Africa 1293insG founder mutation
29
- No fetal involvement
- Moderate hypotonia & oculobulbar involvement
- Disease course: Mild & Stable
- Treatment: Good response to cholinesterase inhibitors
Delta subunits of AChR (CHRND)
● Cholinergic receptor, nicotinic, delta (δ) polypeptide (CHRND)
- Genetics
- Mutation types: Point; Missense & Truncating
- Allelic disorders
- Fast channel syndromes
- Reduced expression of AChRs with fast channel properties
- δ subunit mutation (P250Q): Recessive
- δ subunit mutation (L42P): Fast channel (Short open times); Increased affinity for ACh
- General features: Reduced AChR expression; Fast channel syndromes
- Fast channel syndrome with arthrogryposis: Recessive
- Reduced expression of AChRs with fast channel properties
- Slow channel syndrome: δS268F; Dominant
- Disruption of AChR clustering
- Nonsense mutations
- Multiple pterygium syndrome: Lethal; Recessive
- Fast channel syndromes
- CHRND variant: Disruption of AChR clustering
26
- Genetics
- Inheritance: Recessive
- Mutations: E381K + null mutation
- CHRND mutant protein: Reduced binding to rapsyn
- Clinical
- Early onset: Feeding difficulties
- Ptosis
- Weakness: Moderate, General, Proximal > Distal
- Recurrent episodes of respiratory insufficiency: Provoked by infections
- Treatment: Moderate response to anticholinesterase treatment
- Electrodiagnostic
- Decrement on 3 Hz stimulation
- Genetics
Gamma subunit of AChR
● Cholinergic receptor, nicotinic, gamma (γ) polypeptide (CHRNG)
- CHRNG protein: Expressed in fetal, but not adult, muscle
- Allelic disorders
- Multiple pterygium syndrome (Escobar variant; EVMPS)
- Mutations: Missense & Stop
- Multiple pterygium syndrome, Lethal form (LMPS)
- Mutations: Stop
- Multiple pterygium syndrome (Escobar variant; EVMPS)
Also see
Return to Myopathy & NMJ Index
Return to Acetylcholine & AChRs
Return to Syndrome Index
Return to Neuromuscular Home Page
References
1. J Neurol Sci 1999;166:126-130
2. Lancet Neurol 2015;14:420-434, Brain 2023 Sep 18, Curr Opin Neurol 2024 Jul 23
3. Neurology 2000;54 S3:A138-A139
4. Neurology 2000;54 S3:A138
5. JNNP 2001;70:212-217, Clin Neurol Neurosurg 2002;104:359-363, Journal of Neuroimmunology 2009;217:90–94
6. Neurology 2000;56:A232-A233, Neurology 2002;59:1881–1888
7. J Clin Invest 2001;108:125-130
8. Am J Human Genet 2002;70:875–885, Hum Molec Genet 2003;12:739–748, Neurology 2009;73:228–235
9. Neurology 2002;59:162-168
10. Neuromuscular Disorders 2002;12:547-553
11. Neuromuscular Disorders 2002;12:964–969
12. JNNP 2002;73:766–768
13. Neurology 2002;59:1773–1775
14. Neurology 2002;58:1563–1565
15. J Clin Invest 2003;111:497–505
16. Neurology 2001;57:279–289
17. J Neurol 2003;250:698–701
18. Arch Neurol 2003;60:761-763
19. PNAS 2003;100:7377–7382, Neurology 2019 Mar 1
20. Ann Neurol 2004;55:347–352
21. Hum Molec Genet 2004;13:3229-3240, Neurology 2009;73:1926-1928
22. Muscle Nerve 2005; Online March
23. J Neuroimmunol 2005; Online May, Neurology 2017 Mar 1
24. Neurology 2005;65:144-146
25. Science 2006; Online Aug 17, Brain 2007 Apr 17, Ann Neurol 2008; Online July 14, Neuromuscular Disorders 2012; Online August
26. Brain 2006: Online Aug 17
27. Neurology 2007 Nov 21, Front Immunol 2019;10:769
28. Am J Hum Genet 2008; Online November
29. Neurology 2008;71:1967-1972
30. J Med Genet 2009;46:203-208
31. Am J Hum Genet 2009; Online July, Front Neurol 2020;11:239, Neuromuscul Disord 2025;49:105342
32. Am J Human Genet 2011;88:162–172, Neurology 2013 Jun 21, J Neurol 2017 Jul 15, Brain Behav 2022 Jan 3;e2469
33. Neuromuscular Disorders 2011; Online September
34. Am J Hum Genet 2012; Online June, J Neuromuscul Dis 2023 Mar 28, Neuropathol Appl Neurobiol 2023;Dec 20:e12952
35. Neurology 2012;79:342–347
36. Clin Neurol Neurosurg 2012 Dec 19
37. Brain 2013;136:944-956, Neurology 2017 Jul 21
38. Human Mol Genet 2014;23:1856-68
39. Neurology 2014 ;82:1254-1260, Genet Med 2017 Jul 20, Pediatr Neurol 2026;176:68-76
40. Am J Hum Genet 2014;95:332-339, Neurology 2015; Online October, Muscle Nerve 2021 May 26, J Peripher Nerv Syst 2021;26:237, J Clin Neuromuscul Dis 2025;27:45-56
41. Neurology 2014; Online Nov, Cold Spring Harb Mol Case Stud 2022 Nov 15
42. PLoS One. 2015 Sep 1;10(9):e0137019
43. Am J Hum Genet 2015; Online November, Brain 2019 May 13
44. Brain 2016; Online June
45. Neurology Genetics 2016; Online September
46. Neurology 2016 Sep 2, Neurology 2017 Feb 10
47. Eur J Hum Genet 2016 Dec 14
48. Am J Med Genet A 2017;173:2240-2245, Ann N Y Acad Sci 2018 Jan 28
49. Mol Genet Genomic Med 2018 Feb 14
50. Proc Natl Acad Sci U S A 2019 Sep 30
51. Neuropathol Appl Neurobiol 2020 Apr 8
52. Experimental Neurology 2020; June
53. Am J Med Genet A 2020; Aug 10, Neurol Genet 2020;6(6):e534
54. Clin Genet 2020;97:634-638' Clin Dysmorphol 2025 Jan 10
55. Neurol Ther 2022 May 5
56. Proc Natl Acad Sci U S A 2022;119:e2108672119
57. Neurology 2020;95:e2781-e2793
58. J Neuromuscul Dis 2024 Jul 4
59. Neurol Clin Pract 2024;14:e200277
60. J Neurol 2025;272:498
1/6/2026