- General definition
- MG with any pattern of: Pupil sparing Ophthalmoplegia, ± Ptosis
- Epidemiology
- US & Europe: Adult onset
- Asia: Childhood onset; HLA-Bw46
- African MG: Severe EOM dysfunction
- Association
- Decay-accelerating factor (DAF; CD55)
 (c.-198C>G)
9
(c.-198C>G)
9
- Frequency in MG
- Ocular onset of MG in 30% to 50%
- No relation to age or sex
- May occur in patients with, or without, anti-AChR antibodies
- Clinical
- Onset
- Diplopia & Ptosis
- Painless
- Ptosis as only sign: 10% of Ocular MG
- Weakness
- EOM paresis
- Symptomatic diplopia
- Distribution: Usually asymmetric & bilateral
- Fluctuates
- Worse: In evening; With sustained gaxe
- Differs from thyroid eye disease: Worse in morning
- EOM often dysconjugate
- Saccades: Slower at end of eye movement
- Selective patterns of weakness
- Typical: Bilateral; Multiple muscles
- MLF-like (Bilateral medial rectus): Often symmmetric
- Elevator muscles
- Superior rectus & Inferior oblique
- Often symmetric
- Superior rectus weakness
- Most common involvement (65%)
- More common with longstanding symptoms
- Single muscles: Rare (12%)
- IV nerve-like (Superior oblique)
- VI nerve-like (Lateral rectus)
- Medial rectus
- Inferior rectus
- Levator palpebrae
- Ptosis
- Unilateral or Bilateral
- Worse side may vary from day to day
- Worse with sustained up gaze
- Improved with cold
- Pseudoretraction: Excessive contraction of normal lid
- Association
- With attempts to compensate for ptosis of opposite eye
- Cause: Bilateral equal innervation (Heringís law)
- Orbicularis oculi
- Orbicularis weakness: > 95% of ocular MG
- "Peek" sign: Partial opening of palpebral fissure
- Ectropion
- Pupils: Normal
- Fatigue
- Levator palpebrae
- Ptosis: More with sustained up-gaze
- Lid twitch (Cogan's lid twitch sign)
- Movement: With down-gaze to up-gaze
- Twitch: Lid elevates excessively & then droops again
- External link: mrcophth
- Extraocular muscles
- Saccadic slowing with increased length
- Rapid small saccades
- Gaze evoked nystagmus after sustained gaze
- Quiver eye movements
- Orbicularis oculi: Afternoon ectropion
- Prognosis of ocular MG
- Disease course: Variable
- May represent initial stage of generalized MG
- Evolves into generalized weakness in 25% to 60%
- Time to generalized weakness
- Mean 9 to 12 months
- Range 5 to 27 months
- Can remain localized to extraocular muscles: 40% to 75%
- MG that remains selectively ocular for 2 years
- Rarely becomes generalized thereafter
- Immunomodulating treatment
- Greatly reduces likelihood of generalization
- Generalized MG may present with selective ocular changes
- Frequency: 50% of generalized MG patients
- 94% progress to develop generalized signs over 1st 2 to 3 years
- Treatment
- Corticosteroids: Best improvement
7
- Immunosuppression: May be useful
- Anti-AChE medications: Little long term benefit in most patients
- Laboratory
- Thymic hyperplasia
- Less common (4%) than in generalized MG (12%)
- Repetitive nerve stimulation
- Facial nerve most sensitive
- General sensitivity: 18% toi 35%
- Specificity: High
- Single fiber EMG
- Sensitivity: Abnormal in 80% to 99% of MG when facial muscles also studied
- False positives: Other neuromuscular disorders; Poor technique
- Anti-AChR antibodies
- General ocular MG: Present in 50% to 70%
- African-Americans: Present in 25%
- Properties vs generalized MG
- Lower titers
- ? Low-affinity
- Bind better to adult AChRs with e subunit
- Anti-MuSK antibodies: Rare
- Anti-AChE antibodies: Common
- Acetylcholinesterase inhibitors
- Outcome assessment: Quantitative measurement of eye movements
- False positives: LEMS; Botulism; Guillain-Barré; ALS; Intracranial neoplasms
- AChRs at NMJs: Reduced in both ocular & systemic muscles in most patients
- Ice pack test
- Cooling may reduce degree of ophthalmoplegia & ptosis
- Specificity: High
- Sensitivity: 50% to 60%: Less with severe ptosis
- Work-up: Other
- Rule out
- Intracranial lesion: Head MRI
- Thyroid ophthalmopathy: Orbit MRI
- Rule out thymic pathology: Chest CT
- Thyroid function testing
- Ptosis differential diagnosis
- Treatment
- Prednisone is most effective
- Start at 5 mg/day
- Increase daily dose by 5 mg each week
- Stop increasing: Symptoms begin to improve, or 30 mg q.d.
- Taper slowly over months when symptoms have resolved
- Maintenance: Low doses of 10 to 15 mg q.o.d.; Usually few side effects
- Anticholinesterase medications: Often not effectve
- Mechanical: Lid crutches; Taping eyeglasses
- External links
|
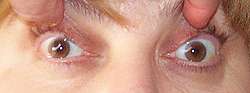
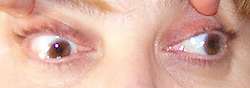
MG: Limitation of adduction
|
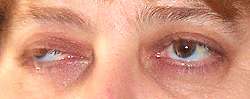
MG: Ptosis
|
Myasthenia gravis:
Eye movements
.avi movie from D Zee MD
|
|