|
Home, Search, Index, Links, Pathology, Molecules, Syndromes, Muscle, NMJ, Nerve, Spinal, Ataxia, Antibody & Biopsy, Patient Info |
FAMILIAL SPINAL CORD SYNDROMES (SPG)
 1
1
|
General principles Protein types Metabolic testing Familial Spastic Paraplegia + Ataxia CNS Dystonia Mitochondrial Motor neuron ALS Primary Lateral Sclerosis Ocular PNS: Motor; PN Systemic Disorders Spasticity: Other syndromes Cerebral palsy Spastic quadriplegic (CPSQ) Leukodystrophies Spastic Ataxias (SPAX) Spine disorders, Familial Spinal cord  From: Wikipedia Adolph Strümpell
|
|
|||||||||
SPG: General Principles 11
|
|

- Protein types
- Endoplasmic reticulum
- Mitochondrial
- Chaperone activity
- Lipid & Ganglioside metabolism
- Golgi
- Myelination
- Axon guidance & Synapse
- Transport proteins
- SPG3A: Atlastin; Dynamin, Vesicle recycling
- SPG4: Spastin; Microtubule cytoskeleton
- SPG10: KIF5A; Anterograde Transport
- SPG11: Spatacsin; Cargo trafficking
- SPG20: Spartin; Late endosomal components
- SPG22: SLC16A2; Iodothyronine transport
- SPG30: ATSV (KIF1A); Synaptic vesicle transport
- SPG 53: VPS37a; Endosomal sorting
- SPG58: KIF1C; Axon transport, Anterograde
- SPG59: USP8
- SPG60: WDR48
- SPG 88: KPNA3; Nuclear-Cytoplasmic transport
- ALS2 & IAHSP: Alsin; ? Vesicle transport
- BICD2: Axon transport
- Nucleotide metabolism, Purine
- Other
- SPG 44: GJA12; Gap junctions
- SPG 61: ARL6IP1; Endoplasmic reticulum
- SPG 70: MARS; ARS, Cytoplasmic
- SPG 71: ZFR
- SPG 77: FARS2; ARS, Mitochondrial
- SPG 87: TMEM63C; Hyperosmolarity-activated channel
- ALDH18A1; Amino acid biosynthesis
- LYST: Membrane fusion & fission
- SINO: KIDINS220; Axon & Dendrite development
- Endolysosome/Autophagy
- SPG 3A: ATL1; BMP signaling, ER-Golgi trafficking
- SPG 4: SPAST; ESCRT-III regulation
- SPG 6: NIPA1; BMP signaling
- SPG8: WASHC5; Endocytosis, Endosomal tubulation
- SPG 10: KIF5A; Axon transport, Autophagic flux
- SPG 11: Spatacsin; Lysosome membrane recycling, Ganglioside clearance
- SPG 15: Spastizin; Autophagosome maturation
- SPG 20: Spartin; ESCRT-III regulation; Endosomal trafficking
- SPG 30: KIF1A; Autophagosome biogenesis
- SPG 39: PNPLA6; BMP signaling
- SPG 42: SLC33A1; BMP signaling
- SPG 47: AP4B1; Autophagosome biogenesis, AP-4 sorting
- SPG 48: AP5Z1; Endolysosomal processing,
- SPG 49: TECPR2; AutophagosomeŚLysosome fusion
- SPG 50: AP4M1; Autophagosome biogenesis, ATG9A sorting
- SPG 51: AP4E1; Autophagosome biogenesis, ATG9A sorting
- SPG 52: AP4S1; Autophagosome biogenesis, ATG9A sorting
- SPG 53: VPS37A; ESCRT-I subunit, Endosomal sorting
- SPG 58: KIF1C: Golgi-ER transport
- SPG 69: Rab3GAP2; Autophagy
- SPG 78: ATP13A2; Autophagy
- SPG 80: UBAP1; ESCRT-I subunit, Endosomal sorting
- SPG 85: RNF170; E3 ubiquitin ligase
- SPG 89: AMFR; ER-associated degradation (ERAD)
- TPP1: Lysosomal exopeptidase
- VPS53: GARP complex
- VCP: Autophagy
Familial Spastic Paraplegia (Strumpell; FSP): Dominant
- SPG3A (FSP1)
19
● Atlastin (ATL1; SPG3A); Chromosome 14q22.1; Dominant or de novo
- Epidemiology: Common cause of dominant, early onset FSP
- Genetics
- ATL1 Gene mutations
- > 500 described
- Most families have private mutations
- Missense (90%)
- Arg217Asp; Arg239Cys (Exon 7); His258Arg (Exon 8); Ser259Tyr (Exon 8); R495W (Exon 12)
- Early onset: Thr156Ile (Exon 4)
- Frameshift: 1688insA (Exon 12)
- Common locations: Exons 7, 8, 12, 13
- Complex SPG phenotype: de novo mutations; Middle domain & Linker regions
- Allelic with: Hereditary Sensory Neuropathy ID
- ATL3 mutations: Hereditary sensory neuropathy with pedal bone destruction
- ATL1 Gene mutations
- Atlastin protein
- Homology
- Dynamin family of large GTPases: Dynamins play role in
- Synaptic vesicles: Recycling
- Mitochondria: Maintenance & Distribution
- Guanylate binding protein 1 (GBP1)
- Dynamin family of large GTPases: Dynamins play role in
- Expression: Predominantly in CNS
- Enriched in cerebral cortex: Especially lamina V
- Subcellular: Golgi apparatus; Oligomeric integral membrane protein
- Mediates formation of tubular ER network
- Interacts with: ARL6IP1
- Homology
- Clinical
- Onset
- Age: Early
- Range: 1 year to 7th decade
- Mean in 1st or 2nd decade
- Anticipation & variation within families
- Spasticity
- Age: Early
- Spastic paraparesis
- Legs
- Motor dysfunction
- Atrophy: Distal legs
- Bladder dysfunction: Some patients
- Tendon reflexes: Increased in legs > Arms
- Sensory loss
- Mild
- Vibration sense
- Course
- Progression: Static or Very slow
- Few with loss of walking: ? Related to contractures
- CNS other: Occasional patients
- Neurodevelopmental delay
- Bulbar symptoms
- Upper limb spasticity
- Ataxia
- Seizures
- MRI: Hyperintensities
- Mutations: Middle domain & Linker regions
- Onset
- Comparison to SPG4
- Onset: Earlier
- Course: More benign
- Laboratory
- Electrodiagnostic: Sensory-motor axonal neuropathy (R495W mutation)
- Pathology: Predominant loss of large myelinated axons
- SPG3A variant syndrome: Hereditary Sensory Neuropathy I
(HSN 1D)
81
- Genetics
- Atlastin mutations: Asn355Lys; Glu66Gln; c.976 delG [p.Val326TrpfsX8]
- Inheritance: Dominant
- Clinical
- Variable within families
- Some patients with spastic paraparesis & no neuropathy
- Onset
- Age: Early adult
- Trophic changes: Skin & Nail
- Childhood spasticity: In one patient
- Sensory loss
- Severe
- Distal
- Weakness
- Distal
- Legs
- Acromutilation
- Osteomyelitis
- Amputations: Toe & Foot
- Pain
- Lancinating
- Some patients
- Tendon reflexes
- Knee: Increased or Normal
- Ankles: Increased or Reduced
- Variable within families
- Laboratory
- Nerve conduction testing: Axonal loss in most but not all
- Nerve biopsy: Axon loss
- Genetics
- SPG4 (FSP2)
● Spastin (SPAST);
Chromosome 2p22.3; Dominant
- Epidemiology
- Most common dominant FSP: 28% to 50% of families
- Prominent variation of severity, even within families
- Similar clinical phenotype with missense & truncation mutations
- Male ≥ Female
- ? Anticipation in some families: Probably related to very variable phenotype
- Genetics
- Mutations
- > 250 different pathogenic mutations identified 9
- Patterns
- Types: Deletions; Insertions; Base substitutions
- Functional types: Missense; Nonsense; Splice-site; Deletion & Insertion
- Locations
- Many exons & splice sites
- Missense commonly in functional AAA domain
- Some truncation mutations
- Clustering in some domains
- AAA (ATPases associated with diverse cellular activities)
- MIT (microtubule interacting and trafficking)
- MTBD (microtubule-binding domain)
- N-terminal region (228-269 residues)
- Commonly private: Specific mutation unique to most individual kindreds
- Missense: May have earlier onset
- Exon 10-12 duplication
- Brazilian family
- Males: Earlier age of onset; More severely affected
- Females: 50% unaffected
- Large exon deletions
- Onset age: Earlier
- Clinical syndrome: Similar
- Frequency: Common
- Technical: May not be detected by gene sequencing
- Large deletion involving SPAST & neighboring DPY30
gene
- Spastic paraplegia: Early childhood onset
- Other systems: Cognitive & bladdder involvement
- Females: Miscarriages
- 2 mutations
- Compound heterozygote
47
- Mutations: 2 Missense; P361L & S44L
- More severe disease
- Infantile onset
- Parents: Asymptomatic or Paraparesis
- Homozygous mutation: S44L
- Adult onset
- Paraparesis
- Homozygous mutation: S545X
154
- Onset: Early childhood
- Spasticity
- Seizures
- Psychomotor delay
- Skeletal: Dysostotic
- Visual disturbances
- Course: Rapid progression
- MRI: Ventricular dilatation
- Compound heterozygote
47
- Disease mechanisms
- Loss of function vs Dominant negative
- Some leaky mutations at splice sites: May explain clinical variability
- Deletions: Earlier onset but similar phenotype vs missense mutations
- No correlation between mutation site & disease severity
- Gly563Ala polymorphism in HSP60: May cause earlier onset of SPG4
- Mutations
- Protein
- ATPase family:
ATPases Associated
with diverse cellular Activities (AAA)
- Share 230 amino acid domain with ATPase function
- Subfamily-7: Meiotic group
- Expression: Early & ubiquitously in fetal and adult tissues
- Subcellular Location: Perinuclear; Punctate
- Structure
- 2 Leucine-zipper domains & 1 Coiled-coil dimerization motif
- Homology with 26S proteasome subunits
- Interacts with: RTN2
- Functions
22
- Associates with microtubule cytoskeleton
- Binding via N-terminal region
- Regulation by ATPase activity of AAA domain
- Promotes microtubule disassembly: Action similar to microtubule-severing protein, katanin

- ? Involved in
- Fine regulation of microtubule cytoskeleton
- Assembly or function of nuclear protein complexes
- Chaperone activity: Also see SPG7 & SPG13
- Axon transport
- Mutant protein
- Disappearance of aster
- Formation of thick perinuclear microtubule bundles
- Altered ability to regulate interaction with tubules
- Constitutive binding to microtubules
- Aggregation of microtubules in long axons of pyramidal neurons
- ? Disrupts ATPase activity of the AAA cassette
- Forms aggregates: Normal protein does not form aggregates
- Associates with microtubule cytoskeleton
- ATPase family:
ATPases Associated
with diverse cellular Activities (AAA)
- Clinical
- Onset
- Typical
- Range: 0 to 74 years; Variable within & between families
- Signs: Leg stiffness (86%); Gait unsteadiness (19%)
- Penetrance (+ Examination)
- 20 years: 60%
- 40 years: 85%
- Asymptomatic patients (Non-penetrance): 6% at age 70
- Sub-clinical manifestations: ~ 20% of patients with + exam are unaware of symptoms
- Spasticity (100%)
- Scissoring gait
- Extensor plantar responses (88%)
- Tone increased
- Muscle spasms & leg cramps
- Legs (94%) > Arms (22%)
- Weakness: Legs; 70%
- Sensory: Vibration reduced in legs (30% to 50%)
- Tendon reflexes: Increased at knee and ankle
- Neurogenic bladder: Common (34%), not universal
- Cognitive impairment: 40%
- Subcortical
- Frequency: Higher with increasing age
- Not related to severity of paraplegia
- Late: Back pain (50%)
- Progression: Faster in older onset patients
- Adult onset
- Slowly progressive spastic paraparesis with bladder dysfunction
- Functional deficit: Mild in most; Severe in < 20%
- "Congenital" onset: May be non-progressive
- Adult onset
- Onset
- Lab
38
- MRI: Normal
- Nerve conduction studies: Normal
- Central motor conduction times: Normal
- Pathology
- Spinal cord: Predominant loss of large myelinated axons
- Cortex
- Neuronal loss
- Hippocampus: Tau reactive neurofibrillary tangles
- Limbic & Neocortex: Tau reactive balloon cells
- Substantia nigra: Lewy bodies
- Epidemiology
- SPG6 (FSP3)
41
● Nonimprinted in Prader-Willi/Angelman syndrome (NIPA1)
; Chromosome 15q11.2; Dominant or de novo
- Epidemiology: > 100 patients
- Genetics
- Mutations
- Missense
- Thr45Arg
- Gly106Arg (316G>A & 316G>C): Hot spot; Complex phenotypes common
- c.159C>G, c.298GC>A, c.731AC>G, c.748AC>C): Pure phenotype common
- Mechanism: Probably dominant negative
- Missense
- Prader-Willi region
- Gene probably commonly deleted in Prader-Willi & Angelman syndromes
- Other Prader-Willi gene: MAGEL2 (Lethal congenital contractures)
- Sporadic ALS: Copy number variation
- Mutations
- NIPA1 protein
- Location: Probably membrane; ? Transporter or Receptor
- 9 predicted membrane spanning regions
- Constitutively expressed in all tissues: Enriched in nervous system
- Clinical
- Onset: Mean 22 years; Range 2 to 35 years
- No anticipation
- Spastic paraparesis
- Relatively severe
- Pure syndrome: 80%
- Tendon reflexes: Brisk in Arms & Legs
- Extensor plantar responses
- Bladder dysfunction (10%)
- Weakness & Atrophy: Distal in legs
- Sensory loss: Vibratory in legs; Frequent; Mild
- Pes Cavus: Common
- CNS: Epilepsies, generalized, in some patients ± treatment responsive
- Polyneuropathy: 6%
- Course
- Progression: Relatively rapid paraparesis
- May need wheelchair in 30's to 50's
- SPG8
61
● WASHC5 (KIAA0196, Strumpellin)
; Chromosome 8q24.13; Dominant
- Epidemiology: 6 families - North American, British, Brazilian, Canadian
- KIAA0196 gene mutations
- Missense: Val626Phe (Hotspot); Leu619Phe; N471D
- Impair protein function
- Increased copy number associated with prostate cancer
- Strumpellin protein: Coded by KIAA0196 gene
- α-helical protein
- Contains spectrin repeat domain
- Expression: Ubiquitous
- Knockout: Impaired motor neuron outgrowth in spinal cord
- Clinical
- General
- Pure spastic paraplegia
- Intrafamilial phenotypic heterogeneity: Present
- Onset age: 22 to 60 years
- Pyramidal signs
- Spasticity
- Hyperreflexia
- Extensor plantar responses
- Gait disorder
- Relatively severe: 60% use wheelchair by age 40
- Spasticity
- Weakness: Hip flexion; Ankle dorsiflexion
- Gait: Spastic diplegia
- Sensory loss: Vibratory in feet
- Bladder disorders (50%): Urgency; Incontinence
- Pes cavus
- Occasional: Dysmetria (Finger-Nose)
- General
- Muscle biopsy: Normal
- SPG10
6
● Kinesin Heavy Chain 5A (KIF5A); Chromosome 12q13.3; Dominant
- Epidemiology: 3% of European AD HSP families
- Genetics
- Mutations
- Types
- Missense: R162W; Asn256Ser; Arg280Cys; Tyr276Cys; Ala361Val
- Other: In-frame deletion; Splice site
- Locations
- Often in kinesin motor or neck domain of kinesin
- Hot spots: R204, N256, R280
- Types
- Allelic disorders
- CMT2 syndrome
- ALS25
- ALS sporadic: Pro986Leu; Faster disease progression 150
- ? LHON, Dominant, Paraneoplastic
- Myoclonus, intractable, neonatal (NEIMY)
- Mutations
- KIF5A protein
- KIF5
- 3 heavy chain isoforms
- KIF5A: Neuron specific
- KIF5B: Ubiquitous
- KIF5C: Neuron specific
- Form: Dimers; Complex with kinesin light chains
- Anterograde axon transport
- Transport many cargos by binding to adaptor proteins
- 3 heavy chain isoforms
- Mediates transport of
- RNA & RNA-binding protein granules
- VAPB
- Mitochondria
- Mutation effects
- Prevents stimulation of the motor ATPase by microtubule-binding
- Impairs axonal transport
- Reduces gross cargo flux: Leads to deficient supply of synapse
- KIF5
- Kinesin disorders
123:
Anterograde axon transport machinery
- Kinesin 1
- Kinesin 3
- KIF1A (ATSV): SPG30; HSMN with ulcero-mutilation (HSAN 2C)
- KIF1C: SPAX2/SPG58
- KIF14
: MKS12
; MCPH20
- KIF 16A (STARD9)
: Microcephaly & Blindness
- Kinesin 4
- Kinesin 5
- KIF11
:
Microcephaly with or without chorioretinopathy, lymphedema or mental retardation (MCLMR)
- KIF11
- Kinesin 7
- KIF10 (CENPE)
: Primary microcephaly 13 (MCPH13)
- KIF10 (CENPE)
- Kinesin 9
- KIF6
: Intellectual disability
- KIF6
- Kinesin 12
- KIF15
: Microcephally & Thrombocytopenia
- KIF15
- Kinesin 13
- KIF2A
: Cortical dysplasia, complex, with other brain malformations 3 (CDCBM3)
- KIF2A
- KIF1Bβ: ?? CMT 2A; Multiple sclerosis predisposition
- KIF1B: ?? CMT2A1
- KIF26B: Ponto-Cerebellar Atrophy with SMA
- KLC2: SPOAN
- KLC4: CONDRHN
- Kinesin-binding protein (KIFBP; KIF1BP/KBP
)
- Goldberg¢Shprintzen syndrome (GOSHS)
- Goldberg¢Shprintzen syndrome (GOSHS)
- Paclitaxel toxicity
- Clinical
- Onset
- Age
- Mean 10 to 23 years
- Range: Infancy to 55 years
- Varies within families
- Gait disorder
- Age
- Pure spastic paraparesis: Some patients
- Spastic paraparesis
- Hyperreflexia: Lower limbs
- Extensor plantar responses
- Disability (Wheelchair): 23%
- Cerebellar signs in arms: 23%
- Foot deformity: 92%
- Bladder dysfunction: 62%
- Sensory loss: Vibration 31%
- Amyotrophy: Rare
- Progression
- Moderate
- Variable severity: From asymptomatic to wheelchair at 40 years
- Most patients continue to ambulate independently
- Spastic paraparesis
- Complex phenotypes: May include
- Peripheral neuropathy
- Sensory-Motor
- Axon Loss
- Upper limb amyotrophy: Severe; Silver syndrome-like
- Mental impairment
- Parkinsonism: Tremor; Bradykinesia; Rigidity
- Deafness
- Retinitis pigmentosa
- Varicose veins
- DupuytrenÆs disease
- Frequency: 10% of complicated forms of SPG
- Peripheral neuropathy
- Onset
- Laboratory
- Electrophysiology
- Subclinical sensory-motor neuropathy in all patients
- Electrophysiology
- KIF5A Variant syndrome: CMT2 ± Pyramidal signs
98
- Epidemiology: 3 patients
- KIF5A mutations: D232N; R280C; R280H; Leu558Pro
- Clinical
- Onset age: 8 to 42 years
- Polyneuropathy
- Weakness
- Distal > Proximal
- Legs > Arms
- Symmetric
- Muscle wasting: Distal
- Sensory loss
- Pin & Vibration
- Weakness
- Pyramidal syndrome (67%)
- Spasticity
- Gait disorder
- Tendon reflexes: Increased
- Course: Progressive to Cane or Wheelchair
- Skeletal: Pes cavus
- Laboratory
- Nerve pathology
- Axon loss
- Axon regeneration
- NCV: Axonal neuropathy
- SNAP & CMAP amplitudes: Reduced or absent, Legs > Arms
- Velocities: 40 to 52 M/s
- Nerve pathology
- Also see: CMT + Upper motor neuron
- KIF5A Variant syndrome: ALS 25
118
- Epidemiology: 0.5% of FALS; 9 families
- Genetics
- Mutations
- Location: C-terminal
- Often splice site: Also frameshift & missense (Arg1007)
- Splicing of exon 27: Commonly affected
- Mutation effects
143
- ? More penetrance with: Loss of function
- Common
- Exon 27: Skipping
- Tail domain altered
- Defective autoinhibition: Gain-of-function 135
- Cytoplasmic inclusions
- Neuromuscular junctions: Abnormal
- RBM24: More cytoplasmic location
- Inheritance: Dominant or Sporadic
- Mutations
- Clinical
- Onset age
- Adult
- Range: 29 to 56 years
- Younger than typical ALS
- Weakness
- Asymmetric
- Arms & Legs
- Bulbar
- Upper motor neuron
- Emotional lability
- Course
- Progressive: Slower than typical ALS
- Death in 3 to > 10 years
- Onset age
● Reticulon 2 (RTN2)
- Epidemiology: USA, Welsh, German & Italian families
- Genetics
- Mutations: Deletion; Frameshift & Truncation
- Disease mechanism: Haploinsufficiency
- Some heterozygous mutations present without disease
- Allelic disorder
- Reticulon 2 protein
- Clinical
- Onset
- Age: Mean 7 to 14 years; Range 5 to 39 years
- Spinal
- Spasticity: Legs > Arms
- Reflexes
- Tendon: Legs increased
- Plantar: Extensor
- Sensory loss: Vibration (11% to 80%)
- Bladder dysfunction: 33%
- Progression: Slow
- Functional deficit
- Mild in some
- Many require wheelchair within 4 years of onset
- Other
- Cerebellar signs: 12%
- Systemic disorders: None
- Foot deformity: 22%
- Onset
- Laboratory
- Nerve conduction & EMG: Normal
- MRI: Spinal cord usually normal
- SSEP: Delayed
- RTN2 variant: HMNR11, Distal motor neuropathy + Lower limb spasticity, Recessive
145
- Epidemiology: 7 families; 14 patients
- Genetics
- Inheritance: Recessive; Homozygous
- Mutations: Loss of function; Most stop
- Clinical
- Onset age: 1 to 6 years
- Weakness
- Distal
- Arms & Legs
- Finger extensors
- Lower limbs
- Spasticity
- Tendon reflexes: Increased
- Sensation: Normal
- Course
- Slow progression
- Remain ambulatory
- Laboratory
- NCV: Motor neuropathy
● Heat-shock 60-kD protein 1 (HSPD1; HSP60)
- Epidemiology: 2 families; Unusual cause of FSP
- Genetics
- Mutations: Missense (Val72Ileu); Deletion (Exons 4 to 17)
- Multiple pseudogenes in addition to functional gene
- Gly563Ala polymorphism: May cause earlier onset of FSP with spastin mutations (SPG4)
- Allelic with: Hypomyelination & Leukodystrophy syndrome
- HSPD1 protein
- Chaperonin: Involved in folding & assembly of proteins
- Location
- Most protein: Mitochondrial matrix
- 20% Extramitochondrial
- Mutant protein: Inactive
- Other FSP with chaperone protein mutations: SPG4; SPG7
- Spasticity & Hyperreflexia
- Lower (67%) & Upper limbs (67%)
- Extensor plantar responses (56%)
- Sensory loss: Vibration (77%)
- Bladder dysfunction (14%)
- Functional deficit: Severe in 40% to 50%
- Systemic disorders: None
- Nosology
- Pelizaeus-Merzbacher¢Like Disease (PMLD)
- Hypomyelinating leukodystrophy 4 (HLD4)
- Epidemiology: Israeli Bedouin kindred, 23 children
- Genetics
- Clinical
- Intra-familial variation
- Onset: Early
- Hypotonia: Poor head control; titubation
- Eye: Nystagmus, rotary; Strabismus
- Psychomotor developmental delay & regression
- Spasticity: More prominent with survival > 2 years
- Head circumference: Decreased growth rate
- Seizures: 60%
- Feeding problems: Malnutrition & Growth failure
- Course
- Exacerbation during fever
- Progression: Rapid (Death < 2 years to slow
- Death: 1st two decades; Aspiration pneumonia; Sudden death
- Laboratory
- MRI: Hypomyelinating leukodystrophy: No normal myelination
- Urinary ethylmalonic acid: High on 60%
- Plasma lactate: High during acute illness
- CSF lactate: Normal
- Muscle biopsy
- Electron microscopy: Abnormal 25%
- Cytochrome c oxidase activity: Mildly reduced 25%
- Other hypomyelination & leukodystrophy syndromes
- Encephalomyopathy: COX6B1
- Pelizaeus-Merzbacher: PLP
- PMLD: GJA12
- Cockayne syndromes
- Hypomyelination, Atrophy of basal ganglia & cerebellum
- Hypomyelination & Congenital cataract syndrome (HCC): DRCTNNB1A
- Leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation (LBSL): DARS2
- Vanishing white matter
● Chromosome 9q33-q34; Dominant
- Genetic: Similar locus to ALS4; HMN + Upper motor neuron signs
- Epidemiology: Italian family
- Onset
- Age: Mean 47 years; Range 36 to 55 years
- Clinical: Spastic gait disorder; Bladder dysfunction
- Some patients unaware of signs
- Clinical
- Spastic paraparesis
- Spasticity: Legs
- Hyperreflexia: Legs 100%; Arms 20%
- Extensor plantar responses (90%)
- Bladder dysfunction (100%)
- Sensory loss: Reduced vibration in 40%
- Functional deficit: Mild in most; Wheelchair in 10%
- Skeletal: Scoliosis (20%)
- Systemic disorders: None
- Spastic paraparesis
- Laboratory
- Motor evoked potentials: Slowed central motor conduction velocity in legs
● Chromosome 1p31.1-p21.1; Dominant
- Epidemiology: Scottish family
- Onset
- Age: 2nd & 3rd decade; ? Anticipation
- Clinical: Gait disorder
- Clinical
- Spastic paraparesis
- Spasticity: Legs
- Hyperreflexia: Legs 100%; Arms 10%
- Extensor plantar responses (100%)
- Bladder dysfunction (30%)
- Sensory loss: Reduced vibration in 15%
- Functional deficit: Major gait disorder or wheelchair in 60%
- Paraesophageal hernia: Persistent vomiting (80%)
- Hearing loss: Sensorineural
- Spastic paraparesis
- Laboratory
- Hyperbilirubinemia
- NCV & EMG: Normal
- Motor evoked potentials: Reduced from lower limbs
- CNS imaging: Normal
● Receptor expression¢enhancing protein 1 gene (REEP1)
- Epidemiology
- > 40 families, most European
- Frequency
- 2% to 6% of Dominant spastic paraparesis
- 8% of Pure SPG
- Genetics
- Mutation types: Varied
- Most common: Frameshift; Small insertions, Deletions or Splice site causing premature termination
- Specific examples
- 1 base pair deletion: c.507delC; c.526delG (Gly176fs)
- Splice-site mutation: c.182-2ArG,
- Missense: P19R; Ala20Glu
- UTR changes: c.606.43GrT; c.606.50GrA
- Alter sequence in binding site for microRNA (miRNA) gene miR-140
- Nonsense: Arg113X
- General disease mechanisms
- Haploinsufficiency
- Loss of protein function
- Allelic disorders
- Mutation types: Varied
- REEP1 protein
- Location
- Tubular portion of peripheral endoplasmic reticulum (ER)
- ER-Mitochondria interface
- Expression: Ubiquitous; Neuronal & Non-neuronal tissues
- Functions
- Rab-mediated vesicle transport
- Facilitates ER-mitochondria interactions
- ? Involved in chaperone-like activities
- Location
- Clinical
- Common phenotype: Pure spastic paraplegia
- Onset age
- Bimodal
- 1st & 2nd decade (> 3 years): 71%
- 31 to 91 years
- Inter- & Intrafamilial variation
- Penetrance: Incomplete
- Bimodal
- Weakness: Proximal legs; Hands (Mild)
- Upper motor neuron signs
- Spasticity: Legs; Gain
- Plantar response: Extensor
- Ankle clonus: 50%
- Sensation: Normal
- Scoliosis: Mild; 30% of families
- Peripheral neuropathy: Some patients
121
- Axon loss
- Compression
- Carpal tunnel: Common
- Multifocal in a few
- Autonomic: Some with adrenergic dysfunction
- Some patients have
- Amyotrophy: Silver syndrome
- Bulbar palsy: More severe disease
- Mitochondrial dysfunction
- Cerebellar ataxia
- Dementia
- Laboratory
- Nerve conduction: Normal velocity
- MRI: Normal spinal cord & Cranial
- REEP1 Variant syndrome: Hereditary Motor Neuropathy 5B (HMND12; HMN 5B)
86
● Receptor expression¢enhancing protein 1 gene (REEP1); Chromosome 2p11.2; Dominant
- Epidemiology: 1 Austrian family
- Genetics
- Inheritance: Dominant
- REEP1 mutation
- Splice-site: c.304-2A>G
- In-frame deletes exon 5
- Encodes internally shortened protein
- Allelic with: SPG31
- REEP1 protein: Expressed in lower motor neurons
- Clinical
- Onset age: 2nd decade
- Weakness & Muscle atrophy: Intrinsic hands; Distal legs, mild
- Tendon reflexes: Absent at ankles
- Upper motor neuron features: None
- Pes cavus
- Gait: Mild steppage in some patients
- Nerve conduction studies
- CMAP amplitudes: Reduced in arms & legs
- Motor conduction velocities: Normal or mildly reduced
- SNAP amplitudes: Normal
- REEP1 Variant syndrome: Distal spinal muscular atrophy-6, Autosomal recessive (DSMA6)
- Nosology: Axonal neuropathy & Diaphragm palsy, Congenital 104
- Epidemiology: 3 families
- Genetics
- Inheritance: Recessive
- Mutation: Homozygous; Splice donor (c.303+1-7GTAATAT>AC, p.F62Kfs23*; NM_022912)
- Clinical
- Onset age: Congenital
- Hypotonia
- Weakness
- Distal ± Proximal
- Legs > Arms
- Respiratory: Diaphragm palsy
- Skeletal
- Arthrogryposis, Distal
- Palate: High arched
- Tendon reflexes: Increased; Reduced at ankles
- Course: Static or Slow progression
- Laboratory
- NCV: CMAP ± SNAP amplitudes reduced or absent; Velocities normal
- Muscle histology: Fiber type grouping; IM nerves with loss of neurofilaments
- Nerve pathology: Axon loss
- Brain MRI: Normal
- Heterozygous parents: Normal or Mild skeletal changes
- Differential diagnosis: SMARD
● ? ZFYVE27
- Epidemiology: German family
- Genetics
- Mutation associatd with disease is Variant of Unknown Significance (VUS)
- Missense
- G191V (G105V of isoform-c)
- ZFYVE27 protein
- Interacts with spastin
- FYVE-finger family
- Expressed in punctate vesicles
- Overlapping expression with endosomal & endoplasmic reticulum markers
- Mutant protein
- Aberrant intracellular pattern in its tubular structure
- Interaction with spastin severely affected
- Clinical: Pure spastic paraparesis syndrome
- Onset
- Age: Adult
- Pes equinus: Many years before symptoms
- Gait disorder
- Spastic paraparesis
- Spasticity
- Tendon reflexes: Brisk or Normal in legs
- Progression: Slow over decade; Eventual inability to walk
- Sensation: Normal
- Onset
● Chromosome 12q23-q24; Dominant
- Epidemiology: German family
- Onset: Mean 24-30 years; Range 14 to 33 years
- Clinical
- Spastic paraparesis
- Legs > Arms
- Weakness: Legs
- Plantar responses: Extensor
- Tendon reflexes: Brisk
- Sensory loss: Pan-modal; Legs
- Bladder: Urinary urgency
- Amyotrophy: Moderate to severe
- Polyneuropathy: Motor & Sensory
- Progression: May develop walking aid or wheelchair dependency after 1 to 2 decades
- Spastic paraparesis
- Laboratory
- Brain MRI: Normal
- Central motor conduction time: Prolonged
- Motor & Sensory NCV: Abnormal in legs
- NCV: Mildly slow
- Motor DL: Prolonged
- SNAP amplitude: Reduced
● Chromosome 8p21.1-q13.3; Dominant
- Epidemiology: French family
- Genetics
- Possible incomplete penetrance
- ? Phenotype modifier at: 10q22.3-23.31: Polymorphism could lead to more severe phenotype
- Locus near SPG5
- Onset age: 8 to 60 years; Mean 32 years
- Clinical: "Uncomplicated"
- Spastic paraparesis
- Legs > Arms
- Gait disorder: Spastic
- Tendon reflexes
- Brisk in Legs ± Arms (60%)
- Ankle clonus: Some patients
- Plantar response: Extensor
- Bladder dysfunction (38%)
- Sensation: Vibration reduced at ankles (38%)
- Course
- Slowly progressive
- No requirement for walking aids
- Proximal weakness: Legs (Psoas)
- Spastic paraparesis
- Laboratory
- MRI: Normal
- EMG: Normal
- Evoked potentials: Normal
● Chromosome 4p16-p15; Dominant
- Epidemiology: Italian family
- Onset
- Age: Mean 17 years
- Gait disorder: Spastic
- Clinical
- Spastic paraparesis
- Legs & Arms
- Gait disorder: Spastic
- Tendon reflexes: Brisk in Legs
- Plantar response: Extensor
- Sensation: Normal or Reduced at ankles
- Pes cavus
- Course
- Progressive
- Walking aids or Wheelchair
- Weakness
- Hands: Thenar & 1st dorsal Interosseus; Uni- or Bilateral
- Foot: Extensors, Some patients
- Spastic paraparesis
- Laboratory
- CMAP amplitudes: Variably reduced, Median > Ulnar
- EMG: Chronic denervation, Distal
- Evoked potentials: Normal
- Also see: SPG17 (Silver syndrome)
● Chromosome 11p14.1-p11.2; Dominant
- Epidemiology: Chinese family
- Genetics: Linkage with low LOD score (2.36)
- Onset age: 2nd decade; Mean 17 years
- Clinical
- Spastic paraparesis
- Legs > Arms
- Gait disorder: Spastic
- Tendon reflexes: Brisk
- Urinary urgency
- Sensation: Normal
- Course: Slowly progressive
- Weakness
- Legs: Proximal
- Arms: Small hand muscles
- Spastic paraparesis
● Solute carrier family 33 (Acetyl-CoA Transporter), Member 1 (SLC33A1)
- Epidemiology: One Chinese family
- Genetics
- Mutation
- Missense: Ser113Arg
- Mechanism: ? Haploinsufficiency
- Allelic disorders
- CCHLND, Recessive
- Chromosomal duplications of 3q25.31 locus: Autism; Seizures
- Mouse SLC33A1 overexpression: Progeria phenotype
- Mutation
- SLC33A1 protein
- Cytoplasmic: Endoplasmic reticulum
- Copper chaperone
- Acetyl-CoA transporter
- Transports acetyl-CoA from cytosol into ER lumen
- Necessary for O-acetylation of gangliosides
- Acetyl group in Nε-lysine acetylation
- May play role in axon growth
- Clinical: Spastic paraparesis
- Onset Age: 4 to 40 years; Rare incomplete penetrance
- Distribution: Legs
- Tendon reflexes: Increased in legs
- Extensor plantar response: Common
- Disability: Most patients remain walking
- Pes cavus: Some patients
- Laboratory
- MRI: Normal
- EMG: Normal
- Muscle biopsy: Normal
- SLC33A1 variant: Congenital Cataracts, Hearing loss & Neurodegeneration (CCHLND)
- Nosology: Huppke-Brendel syndrome (HBS)
- Epidemiology: 5 families
- Genetics
- Inheritance: Recessive
- Mutations: Missense; Splice; Stop
- Clinical
- Onset age: Infancy
- Cataracts: Congenital
- Psychomotor development: Delayed; Hypotonia, severe
- Eyes: Gaze fixation instability; Rotary nystagmus
- Hearing loss
- Seizures
- Course: Progressive
- Laboratory
- Brain MRI: Cerebral & Cerebellar atrophy; Hypomyelination
- Muscle: Mitochondria abnormal; Cytochrome c oxidase activity reduced
- Copper & Ceruloplasmin: Low in serum
- Neutropenia
● Chromosome 21q22.3; Dominant
- Epidemiology: 1 American family, 8 patients
- Genetics
- Inheritance: Dominant
- Clinical
- Onset
- Age: Mean 45 years; Range 35 to 56 years
- Leg stiffness
- Spasticity
- Legs > Arms
- Tendon reflexes: Brisk in Legs > Arms
- Plantar response: Extensor in 40%
- Gait disorder
- Sensory loss
- Proprioception & Vibration
- With disease progression
- Progression: Slow
- Onset
- Electrodiagnostic
- Axon loss: Sensory & Motor
● Receptor expression-enhancing protein 2 (REEP2)
- Epidemiology: French & Portuguese families
- Genetics
- Dominant mutation: Val36Glu
- Recessive mutations: M1T; Phe72Tyr & Splice site (c.105+3G>T)
- Allelic disorders
- SPG72A
: Dominant
- SPG72B
: Recessive
- SPG72A
- REEP2 protein
- Clinical: Pure SPG phenotype
- Dominant family (SPG72A)
- Onset
- Age: Mean 3.7 years
- Stiff legs
- Spasticity
- Gait disorder
- Mild stiffness at rest
- Plantar response: Extensor
- Tendon reflexes: Increased in legs
- Progression: Slow over decades
- Sphincter disturbance (50%)
- Pes cavus 30%
- Postural tremor (20%)
- Sensory loss (20%): Decreased vibration sense at ankles
- Onset
- Recessive family (SPG72B)
- Onset age: 2 years
- Spasticity
- Gait
- Rest
- Plantar response: Extensor
- Tendon reflexes: Increased in legs
- Sensation: Normal
- Course: Progressive to cane use at 8 to 25 years
- Dominant family (SPG72A)
● Carnitine palmitoyltransferase IC (CPT1C)
- Epidemiology: 7 families
- Genetics
- Mutations: Missense (Arg37Cys); Frame-shift
- CPT1C protein
- Endoplasmic reticulum
- Carnitine long-chain acyltransferase (CPT) family
- Location: Neurons; Hippocampus, Hypothalamus, Amygdala, Cerebellum, Motor cortex, Dorsal root ganglia
- Lipid droplets: Synthesis
- Metabolism: Triacylglycerol & Ceramide
- Gypothalamic control of energy homeostasis
- Cognition
- Hippocampal dendritic spine maturation
- AMPA receptor subunit GluA1 synthesis & trafficking to post-synaptic membrane
- Motor function
- Regulator of survival of cancer cells during metabolic stress
- No enzymatic activity in fatty acid oxidation
- CPT1A & CPT1B
- Locations: Liver, Muscle, Brown adipose tissue
- Gate-keeper enzymes for long-chain fatty acids to
- Enter mitochondria as carnitine
- Undergo β-oxidation
- Clinical
- Onset age: Mean 36 years; Range Infancy to 48 years
- Spastic paraparesis
- Legs: Gait disorder
- Tendon reflexes: Increased
- Plantar response: Extensor
- Urinary dysfunction: Some patients
- Course: Slow progression
- Weakness: Proximal
- Sensory: Vibration loss in feet
- Foot deformities: Some patients
- CNS, Other
- Seizures
- Vision impairment
- Intellectual disability: Development; Cognitive & Behavioral; Psychiatric
- Ataxia
- Laboratory
- NCV: Normal
- Brain imaging: Normal or Cerebellar atrophy
- Sensory evoked potentials: Delayed
● Ubiquitin-Associated Protein (UBAP1)
- Epidemiology: > 90 patients; 1.2% of Dominant HSP
- Genetics
- Mutations
- Truncation: Stop, Out-of-frame duplications
- Locations
- After N-terminal UMA domain (Mediates association with ESCRT-I complex)
- Before C-terminal SOUBA domain (Interaction with ubiquitinated proteins)
- Hotspot: Exon 4
- Genetic Mechanism: Dominant negative
- Mutations
- UBAP1 protein
- Subunit of endosomal sorting complex: Required for transport-1 (ESCRT-I)
- Regulator of vesicular trafficking processes
- Proteasomal degradation of ubiquitinated cell-surface proteins
- EGFR (Epidermal growth factor receptor)
- BST2 (Bone marrow stromal cell antigen)
- EGFR (Epidermal growth factor receptor)
- Lysosomal regulation
- Mutated UBAP1 proteins: Loss of recruitment to damaged lysosomes
- Clinical
- Onset
- Age: Median 8 years; Range 4 to 26 years
- Spastic gait
- Spasticity
- Legs
- Tendon reflexes: Increased
- Gait disorder
- Urinary urgency (37%)
- Course: Progressive; Wheelchair in many
- Some patients
- Cerebellar: Ataxia; Saccadic EOM; Limbs
- Cognitive impairment: Mild
- Sensation: Normal
- Onset
- Laboratory
- Brain imaging: Normal
● KCNA2
- Epidemiology: 2 families (Norwegian/French-Canadian; German), 5 patients
- Genetics
- Mutation: Missense; Arg294His
- Allelic disorders
- Ataxia
- Developmental and epileptic encephalopathy 32 (DEE32)
- KCNA2 protein
- Clinical
- Onset age: 1 to 30 years
- Development: Motor or Global delay in some patients
- Intellectual disability: Mild to Moderate
- Spasticity: Legs > Arms; Gait disorder
- Ataxia: Limb & Gait (1 family)
- Bladder disorder: 1 patient
- Polyneuropathy: 1 older patient
- Course: Progressive
- Laboratory
- Brain MRI: Non-diagnostic
- EEG: Epileptiform discharges (50%)
- KCNA2 variant syndrome: Ataxia + Epilepsy
- Epidemiology: 2 families, 2 patients
- Genetics
- Mutations: Arg294His; Arg297Gln
- Inheritance: Dominant
- Clinical
- Onset age: 15 months to Childhood
- Ataxia: Limb & Trunk
- Epilepsy: Febrile seizures; Myoclonus
- Tremor: Hands
- Intellectual disability
- Treatment: Acetazolamide
- Laboratory
- EEG: Spike-Wave
- Brain MRI: Normal
● Chromosome 2q24-2q31; Dominant
- Epidemiology: Central Ohio, US Family
- Genetics: Highly penetrant
- Clinical
- Onset
- Age: 1 to 51 years; Most often in 1st decade
- Dystonia: Childhood
- Spasticity: Adults
- Spasticity
- Legs > Arms
- Gait
- Tendon reflexes: Increased; Clonus
- Dystonia
- Location: Arms, Legs & Generalized
- Present in most, but not all, patients
- Normal: Cognition, Cerebellar & Bladder function
- Course: Progressive
- Onset
- Laboratory
- CNS MRI: Normal
- Nerve conduction studies: Normal
- Dystonia treatment: Deep pallidal stimulation
- Alzheimer's disease ± Spasticity
37
● Presenilin 1 (PSEN1); Chromosome 14q24.3; Dominant
- Genetics
- Mutation: Deletion of exon 9
- Allelic disorders
- Epidemiology: Finnish, British & Australian families
- Onset age: 4th & 5th decade
- Clinical
- Spastic paraparesis: May precede dementia
- Dementia: Rapidly progressive; May be delayed when preceded by paraparesis
- Progression: Death in < 10 years
- Pathology
- Plaques: Non-neuritic; æCotton woolÆ
- Large, round & eosinophilic
- Immunoreactive for A-β
- No congophilic dense core or plaque-related neuritic pathology
- Neurofibrillary tangles
- Amyloid angiopathy: Throughout cerebral cortex
- Plaques: Non-neuritic; æCotton woolÆ
- Genetics
- SOX10
- Adult-onset Leukodystrophy (ADLD)
- Multiple exostoses and spastic tetraparesis
: 1 family
Familial Spastic Paraplegia (RFSP): Recessive
Spastic Paraplegia: Non-syndromic or Complex
- SPG5A
● CYP7B1; Chromosome 8q12.3; Recessive
39
- Epidemiology: Tunisian, American, Australian & British families
- Genetics
- Mutations: Homozygous; Missense or Nonsense; > 17 identified
- Allelic with: Congenital bile acid synthesis defect
- CYP7B1 protein
- Cytochrome P450 enzyme
- Hydroxylates
- Endogenous substrates
- Carbons 6 & 7 of B ring of oxysterols & steroids
- Expression: Liver, Lung, Kidney, Brain & Reproductive tract
- Function
- Role in metabolism of brain cholesterol
- Conversion of 27-OH-cholesterol to 3β, 7α-diOH-5-cholestinoic acid
- Clinical features: Pure HSP with sensory loss
- Onset age: 1 to 40 years; Median 13 years
- Spasticity: Lower limbs; Gait disorder
- Weakness: Legs
- Sensory loss
- Distribution: Legs
- Modalities: Joint Position; Vibration
- Tendon reflexes: Increased in arms & legs
- Plantar responses: Extensor
- Bladder dysfunction: 66%
- Associated signs: ? 2 families with mild cerebellar features late in course
- Course: Progressive
- Loss of walking independently: Median disease duration 23 years
- Wheelchair dependent: Median 33 years
- Laboratory
- Cortical evoked responses: Abnormal
- Muscle biopsy
- Few SDH-positive muscle fibers
- Normal mitochondrial enzyme analysis
- CYP7B1 substrates: Oxysterols; High in serum & CSF
- 25-OHC, 27-OHC & ratio to total cholesterol
- 27-hydroxycholesterol: Serum levels correlate with disease severity & duration
- Atorvastatin + Chenodeoxycholic acid: Improve metabolic abnormalities
- SPG5B
● Recessive- Epidemiology: Tunisian family
- Genetics
- Clinical: Pure spastic pararplegia
- SPG7 or SPG5C
54
● Paraplegin (SPG7 gene; CMAP); Chromosome 16q24.3; Recessive or Dominant (Few mutations)
- Epidemiology
- Frequency: ~1.5% to 6% of SPG families
- European, Moroccan & Turkish families
- May present as sporadic HSP: Predominant spasticity in legs
- Genetics
- Mutation Types
- Frameshift: Insertion or Deletion
- Missense: A10S; Ala510Val (Common); A572V; G577S; F676L
- Inheritance
- Recessive: Usual
- Dominant (Probable): 9 base pair deletion in exon 11; Missense mutations
- Clinical correlations
- Null mutations: Associated with ataxia
- Optic atrophy: Arg470Gln (Homozygous); Asp411Ala (Heterozygous)
- Allelic disorders
- Ataxia, Spasticity + PEO, Adult onset
- Optic atrophy, Non-syndromic, Dominant: Asp411Ala
- SPG7
- Ataxia, Late onset: Heterozygous A510V mutation 153
- Mutation Types
- Protein
5
- Cell localization: Mitochondria
- Inner mitochondrial membrane
- Hetero-oligomer with ATPase family gene 3-like 2 (AFG3L2): Forms m-AAA protease
- Tissue localization: Numerous; Higher in adult than fetal brain
- Structure: Homologous to yeast mitochondrial ATPases (metalloprotease)
- Function: Mitochondrial AAA-metalloprotease
- Substrate: OPA1
- Mutations: Frameshift
- Cell localization: Mitochondria
- Clinical: Pure spastic paraparesis, or Complicated syndromes (69%)
- Onset
- Age: 2nd to 5th decade
- Presenting feature: Gait change
- Clinical features
- Complex phenotype: More with younger onset age
- Upper motor neuron signs
- Tone: Increased; Legs > Arms
- Gait: Spastic
- Tendon reflexes in arms & legs: Increased
- Plantar response: Extensor
- Strength
- Relatively preserved compared to spasticity: May be normal
- Reduced
- Lower ± Upper extremities
- Proximal > Distal
- ? Distal predominance in some
- Sensation: Vibration sense reduced
- Bladder dysfunction (50%)
- Skeletal (40%): Scoliosis; Pes cavus
- CNS
- Eye
- Ptosis
- Movement
- Vertical gaze palsy
- External ophthalmoplegia
- Optic neuropathy
- Frequency: 40% to 100%
- Imaging: May be abnormal in asymptomatic patients
- May be only sign with dominant Asp411Ala mutation
- Cerebellar (57%)
- Limb ataxia
- Dysarthria
- Nystagmus
- Cervical dystonia
- Intellectual disability
- Eye
- Severe cases: Dysarthria; Dysphagia
- Progression
- Slow over decades
- May eventually need walking aids
- Onset
- Laboratory
- Serum CK: May be elevated
- Muscle pathology
- Histology (More severe involvement): COX- fibers; Some ragged red
- Mitochondrial oxidative enzyme activities in muscle or fibroblasts: Variable
- Complex I & II/III: Often Reduced
- Complex IV: Normal or reduced
- MRI: Cerebellar or cortical atrophy; Spinal cord normal
- EMG & NCV: Normal
- SPG7 variant syndrome: Ataxia + Spasticity & PEO, Adult onset
100
- Genetics
- Inheritance: Recessive
- Mutations: One mutation always Ala510Val
- Paraplegin protein
- Clinical
- Onset age: 1st to 5th decade; Mean 36 years
- Ataxia
- Gait & Trunk: Onset
- Limbs & Dysarthria: With disease progression
- Ocular
- Eye: PEO
- Cerebellar: Slow saccades; Nystagmus
- Pyramidal: With disease progression
- Spasticity: Legs
- Tendon reflexes: Brisk
- Plantar response: Extensor
- Course: Progression over years
- Laboratory
- Brain MRI: Cerebellar atrophy
- Muscle
- COX- muscle fibers
- Coenzyme Q10: Low in 1 patient
- Carriers: Some reported with
- Cerebellum: Signs or Atrophy
- Peripheral neuropathy
- Mouse model: Paraplegin knock-out
- Abnormal mitochondria in nerve terminals
- Distal axonopathy
- Spinal cord & PNS
- Axons: Swelling and degeneration
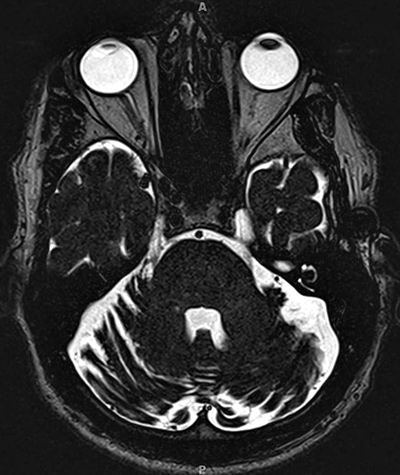
From: R Bucelli
SPG7 variant
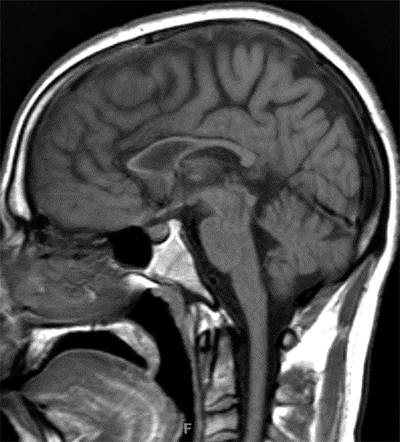
Atrophy of Cerebellar hemispheres +
Medial & Lateral Rectus muscles - Genetics
- Epidemiology
- SPG 11
:
Spastic paraplegia with Thin corpus callosum (ARHSP-TCC)
4
● Spatacsin (ALS5; SPG11; KIAA1840; FLJ21439)
; Chromosome 15q21.1; Recessive
- Epidemiology
- Relatively common: Especially for complex SPG
- North America, Europe, Japan, Turkey
- Genetics
- Mutations: Nonsense, Deletion & Insertion
- Mechanism: Loss of function
- Allelic disorders
- Juvenile ALS, Recessive (ALS5)
- PLS, Juvenile onset
- Parkinsonism, levodopa responsive, juvenile onset
- HMSN, Axonal, Recessive (CMT 2X)
- SPG11
- Spatacsin protein
- Cytosolic & perinuclear distribution: Diffuse & reticular
- Possibly membrane associated
- Adult: Distributed throughout brain
- Neuron location: Axons & Dendrites
- Subcellular: Cytoskeletal & synaptic vesicles; Synaptosomes
- Axon maintenance: Cargo trafficking
- Clinical
- Onset
- Age: Usually before 20 years; Range 1 to 50 years
- Spasticity: Legs
- Mental slowing
- Parkinsonism: Occasional patient; Tremor & Akinesia
- Upper motor neuron signs
- Spasticity: Legs
- Hyperreflexia: Knees; Ankles may be increased or reduced
- Upgoing toes
- Dysarthria: Pseudo-bulbar
- Asymmetry: Mild
- Bladder function: Normal or Abnormal
- Lower motor neuron
- Weakness: Legs
- Wasting: Distal
- Sensory
- Early: Commonly normal
- Later in course: Neuropathy
- Pin, Temperature or Vibration sense reduced
- Neuropathic pain
- Ataxia: Mild
- CNS: Other
- Mental retardation: 25%; Progressive disorder
- Extrapyramidal: Parkinsonism, Juvenile, Levodopa responsive
- Other occasional features
- Eye
- Optic atrophy: May be more prominent on temporal side
- Retinitis pigmentosa
- Cataract
- Movement: Strabismus or Reduced convergence
- Clinodactyly
- Eye
- Course
- Progressive: Gait disorder & disability
- Wheelchair in 3rd decade
- Onset
- Laboratory
- Imaging: White matter lesions
- Corpus callosum
- Agenesis of, or thin, in 25%
- More abnormality: Increasing age; Rostral
- Periventricular
- Cortical atrophy: Frontal
- Corpus callosum
- NCV: Motor neuropathy
- Motor axon loss: Reduced CMAP amplitudes
- SNAPs: Normal
- EMG: Denervation in limbs
- Motor evoked potentials: Central conduction slow
- Imaging: White matter lesions
From: R Bucelli
SPG11: Thin Corpus Callosum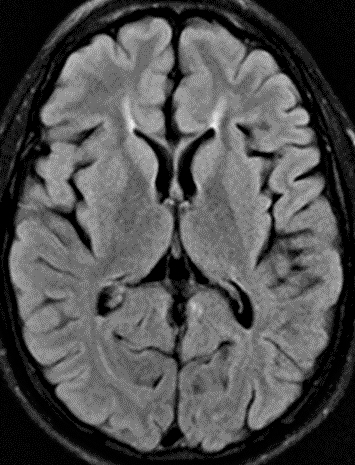
From: R Bucelli
SPG11: Ears of the Lynx sign
(T2 signal projecting from anterior lateral ventricles)- SPG11 variant syndrome: HMSN, Axonal, Recessive (ARCMT2X; CMT 2X)
106
- Epidemiology
- 12 families: 29 individuals
- Italy, Brazil, Canada, England, Iran, Japan
- Genetics
- Mutations: 15 different; Most truncating; Arg945Gly; Some same as for SPG11
- Mutation locations: Entire gene
- Inheritance: Recessive
- Allelic disorders
- Clinical
- Onset age: 4 to 35 years; Mean 11 years
- Weakness
- Distal
- Legs > Arms
- Asymmetric (40%)
- Gait disorder
- Proximal (14%)
- Wasting (Muscle): Legs & Hands
- Spasticity: Some patients
- Sensory loss (85%)
- Legs
- Modalities: Pin; Light touch; Proprioception
- Fasciculations (30%): Legs
- Skeletal
- Pes cavus
- Ankle contractures
- Scoliosis
- Tremor (34%)
- Bladder (31%) or Sexual (24%) dysfunction
- Cognitive impairment (10%)
- Course: Slow progression
- Laboratory
- NCV
- Axon loss: Small CMAP & SNAP amplitudes
- Velocities: Median > 38 m/s
- EMG: Denervation in hands & distal legs
- Nerve biopsy: Loss of Myelinated & Unmyelinated axons; Intra-axon aggregates
- Brain MRI: Thin corpus callosum in some patients
- Serum CK: Normal
- NCV
- Epidemiology
- Epidemiology
- SPG 14
10
● Chromosome 3q27-q28; Recessive- Epidemiology: Italian family
- Onset: Mean 30 years
- Clinical
- Spastic paraplegia
- Spasticity: Leg & gait
- Plantar response: Extensor
- Tendon reflexes: Increased in Legs
- Neuropsychological impairment
- Mental retardation: Mild
- Visual agnosia
- Memory disorder: Short & Long term
- Motor neuropathy
- Distal
- Mild
- NCV: Mild slowing in legs
- Skeletal: Pes cavus
- Progression: Slow over decades
- Spastic paraplegia
- SPG 15
16
● Spastizin (ZFYVE26; KIAA0321)
; Chromosome 14q24.1; Recessive
- Nosology: Kjellin syndrome
- Genetics
- Mutations
- Homozygous
- Truncating: Nonsense; Frameshift deletions; Complex rearrangements
- Mutation effects
- Degradation of mutant RNA or protein
- Loss of C-terminal putative leucine zipper domain & usually FYVE domain
- Mutations
- Spastizin protein
- Onset
- Age: 5 to 23 years
- Gait disorder
- Clinical
- Spasticity & Pyramidal features
- Tetraparesis
- Clonus
- Extensor plantar responses
- Increased tone
- Tendon reflexes: Increased
- Dysarthria
- Fecal & Urinary incontinence: Late in course
- Lower motor neuron
- Weakness: Distal arms & hands; Neck flexion
- Amyotrophy: Distal
- Cerebellar (32%): Mild upper limb ataxia; Some patients
- Cognitive (73%)
- Mental retardation
- Progressive intellectual deterioration in 2nd decade
- Other CNS features in few patients
- Psychosis
- Epilepsy
- Dystonia
- Hearing loss
- Visual acuity decreased
- Pigmented maculopathy
- Mildly reduced visual acuity
- Present at onset or few years later
- Slow progression
- Progression
- Often moderate to severe disability
- Wheelchair in 3 to 21 years
- Spasticity & Pyramidal features
- Laboratory
- MRI: Atrophy of cerebral hemispheres, corpus callosum, brainstem
- NCV: Axonal neuropathy (67%)
- Muscle biopsy: Denervation
- CSF: Normal
- SPG 18B
● Endoplasmic reticulum lipid raft-associated protein 2 (ERLIN2; SPFH2); Chromosome 8p11.23; Recessive
- Epidemiology: 7 families
- ERLIN2 genetics
- Mutations
83: Recessive
- Types: Deletion; Null; Insertion, Out of frame
- Allelic disorders
- SPG18A, Dominant: Spastic paraparesis, Pure
- PLS, Juvenile onset
- Intellectual disability, Motor dysfunction & Joint contractures (IDMDC)
- ALS variant: Onset late in SPG course
- Mutations
83: Recessive
- ERLIN2 protein
- ERAD pathway
- Family: SPFH domain-containing lipid raft-associated proteins
- Location: Lipid rafts of Endoplasmic reticulum (ER)
- Associates with: Activated Inositol 1,4,5-trisphosphate (IP3) receptors
- May act as substrate recognition factor
- Endoplasmic reticulum¢associated degradation pathway of IP3Rs
- Homologous protein: ERLIN1 (SPG62)
- Clinical
- Onset age: 2 to 6 years
- Gait disorder: Spastic
- Spasticity: Legs > Arms
- Weakness: Legs, some patients
- Reflexes: Tendon brisk; Plantar extensor
- Skeletal: Joint contractures; Scoliosis
- CNS: Varies among patients
- Mental retardation: 2 families
- Language disorders
- Epilepsy: 2 families
- Squint
- Course
- Spastic paraparesis: Progressive
- May evolve to: Rapidly progressive ALS-like syndrome
126
- Genetics
- Mutations: V168M & A309V (Dominant); D300V(Recessive); N125S (Sporadic)
- Clinical
- Onset
- Ages: 45 to 66 years
- Disease course before ALS: 20 to 45 years
- Site: Arms or Bulbar
- Other weakness: Respiratory
- Course: Death in 1 to 4 years
- Onset
- Genetics
- Laboratory
- EMG & Muscle biopsy: Normal
- CNS imaging: Often normal; Agenesis or hypoplasia of corpus callosum in 1 family
- ERLIN2 Variant syndrome: Primary Lateral Sclerosis, Juvenile onset
88
- Epidemiology: Saudi family
- ERLIN2 Genetics
- Mutation: Splice acceptor site; c.499-1G>T; Intron 7 of long gene isoform
- Inheritance: Recessive
- Clinical
- Onset
- Age: 8 months
- Difficulty crawling
- Muscle spasms
- Motor functions
- Walking: Delayed
- Progressive loss: After 2 years
- Wheelchair dependent: 11 to 12 years
- Bedridden: 15 years
- Other motor
- Weakness: Distal > Proximal
- Spasticity
- Tongue: Slow movement; No fasciculations
- Tone: Increased
- Tendon reflexes: Increased
- Plantar response: Extensor
- Speech: Progressively lost after 2 years
- Skeletal
- Spine: Kyphosis; Scoliosis
- Palate: High arched
- Upper jaw: Narrow
- Small limbs
- Eye movements: Saccadic pursuit
- Onset
- Laboratory
- Brain MRI: Normal
- NCV & EMG: Normal
- ERLIN2 variant syndrome: SPG18A, Dominant
- Epidemiology: 3 families, 13 patients
- Genetics
- Dominant mutations: SPFH domain
- Mutations: D69V, V71A, S129T, A151V, V168M, A309V
- Clinical: Spastic paraparesis, Pure
- Onset age: 9 to 46 yeaars
- Spastic paraplegia
- Spasticity: Legs
- Gait disorder
- Tendon reflexes: Brisk in legs
- Vibration loss (25%): Legs
- Bladder dysfunction: Few patients
- Course: Slow progression
- No seizures or cognitive Δ
- Laboratory
- Brain MRI: Normal
- NCV: Normal
- SPG, infantile onset (IAHSP)
29
● Alsin (ALS2); Chromosome 2q33.1; Recessive
- Epidemiology: Families in Algeria, Italy & France
- Genetics
- Mutations
- Type: Deletions & Splice site
- Frequency: Found in 4 of 10 families
- Allelic with
- Mutations
- Clinical
- Birth: Normal
- Onset: Infantile
- Spasticity
- Paraplegia: Onset at time of initial walking
- Upper limbs: Onset at 7 to 10 years
- Wheelchair necessary: < 10 years
- Progression to tetraplegia during 2nd decade
- Cranial nerves
- Onset 2nd decade
- Anarthria
- Dysphagia
- Eye movements: Slow
- Survival: Some patients into 3rd & 4th decade
- Cognitive functions: Normal
- Laboratory
- NCV & EMG: Normal
- Muscle biopsy: Normal
- Motor-evoked potentials (MEP): Early loss of corticospinal responses, in contrast with
- Somatosensory-evoked potentials: SSEP: Abnormal only in the later stages
- MRI
- Atrophy: Sylvian, Brainstem & Spinal cord
- Hyperlucencies in T2-weighted images: Along the pyramidal tract in oldest patient
- SPG24: Pure spastic paraplegia
33
● Chromosome 13q14; Recessive- Epidemiology: Saudi Arabian family
- Onset
- 1 year: Standing difficulty
- Gait disorder: Toe walking
- Clinical
- Spasticity: Legs > Arms; Scissor gait
- Tendon reflexes: Brisk in arms & legs
- Speech: Usually normal; May be dysarthric
- Weakness: Mild
- Bladder: Normal
- Sensation: Normal
- Laboratory
- MRI: Normal brain & spine
- SPG 26
50
● β-1,4-N-Acetylgalactosaminyltransferase 1 (B4GALNT1); Chromosome 12q13.3; Recessive
- Epidemiology: > 10 families
- B4GALNT1 genetics
- Mutations: Loss of function; Missense (Arg300Cys; Asp433Ala), Frameshift, Nonsense
- Allelic disorders
- AR-CMT2 + CNS
- Glutaric acidemia, Type 2
: c.263dupG mutation
- Neoplasms: Overexpression promotes progression & metastasis
- B4GALNT1 protein
- Function
- Ganglioside biosynthesis
- Adds GalNAc to carbohydrate moieties
- GA3 → GA2 ganglioside
- GM3 → GM2 ganglioside
- GD3 → GD2 ganglioside
- GT3 → GT2 ganglioside
- Reverse reaction: Performed by β-hexosaminidase
- Other Ganglioside & Glycolipid disorders
- Synthetic
- Infantile epileptic encephalopathy
:
ST3GAL5
- SPG26: B4GALNT1
- Infantile epileptic encephalopathy
- Glycolipid Degradation
- Gaucher disease
: GBA
- Fabry disease
: GLA
- Metachromatic leukodystrophy: ARSA
; Saposin
- Krabbe: GALC
- Tay-Sachs: HEXA
- Tay-Sachs AB variant/GM2 gangliosidosis
: GM2A
- Sandhoff disease
: HEXB
- GM1 gangliosidosis
: GLB1
- Gaucher disease
- Antibodies
- Synthetic
- Function
- Clinical
- Onset ages: 2 to 19 years
- Spastic paraparesis
- Legs: Spastic, moderate to severe
- Arm spasticity (33%)
- Dysarthria, pseudobulbar, mild (47%)
- Tendon reflexes: Brisk in legs; May be reduced late in disease course
- Plantar response: Upgoing in 60%
- Gait disorder: Spastic
- Bladder dysfunction
- Progression: Slow over decades; Most walk independently
- Strength: Leg weakness
- Amyotrophy: Distal Arms & Legs (60%), Late in disease course
- Vibration sense: Reduced at ankles in 50%
- Intellectual
- Impairment: IQ 50 to 70
- Emotional lability
- Cerebellar (55%): Saccadic dysfunction; Nystagmus; Dysmetria
- Pyramidal (44%)
- Skeletal: Pes cavus (50%); Socliosis (67%)
- Tongue: Tremor
- Other occasional
- Dyskinetic facial movements
- Dystonia
- Cataracts
- Strabismus (22%)
- Psychiatric (22%)
- Delayed puberty
- Laboratory
- NCV: Axonal sensory or motor neuropathy (60%)
- EEG: Normal
- MRI: Corpus callosum thick; Cortical atrophy; WM change; May be normal
- Testosterone: Reduced (67%)
- B4GALNT1 variant disorder: AR-CMT2 + Intellectual disability
129
- Epidemiology: 1 Korean patient
- Genetics
- Inheritance: Recessive
- Mutations: c.128dupC; c.451G>A
- Clinical
- Onset: 20 years
- Weakness: Distal legs
- Sensory loss: Large fiber
- Tendon reflexes: Absent at knees & ankles
- Cognitive impairment: Mild to Moderate
- Laboratory
- NCV: SNAP amplitudes reduced
- EMG: Denervation in distal legs
- Brain MRI: Normal
- SPG27: Pure spastic paraplegia
● Chromosome 10q22.1-q24.1; Recessive- Epidemiology: French-Canadian family
- Genetics: Overlap with locus for Dominant SPG9
- Clinical
- Onset age: 25 to 45 years
- Spastic paraparesis
- Legs
- Bladder involvement
- Tongue: Slow movements; Dysarthria
- Progression: Slow; Most walk independently
- Vibration sense: Reduced in feet
- Strength: Normal
- Laboratory
- NCV: Normal
- EMG: Normal; No denervation in legs
- SSEP: Reduced amplitudes
- SPG28: Pure spastic paraplegia
51
● Phospholipase DDHD1
;
Chromosome 14q22.1; Recessive
- Epidemiology: Moroccan, Turkish & French families
- Genetics
- Mutations: Missense; Splice; Stop
- Allelic syndrome: Retinopathy & Neurodegeneration with Brain Iron Accumulation (NBIA)
- DDHD1 protein
- Localization: Cytosol; Microsomes; Mitochondria
- Phospholipase A1, PA preferring
- Tissue expression: Ubiquitous
- ? Maintenance of organelle membranes & intracellular trafficking
- Clinical
- Onset age: Infancy to 15 years
- Spastic paraparesis
- Legs
- Progression: Assisted gait in 5th decade
- Tendon reflexes: Increased in Legs & arms
- Sensory loss
- Vibration: Reduced in distal legs
- Pin: reduced in legs
- Strength: Mild leg weakness
- Eye: Saccadic pursuit
- Laboratory
- Nerve: Axon loss
- Muscle: Mitochondrial changes in some patients
107
- mtDNA deletions
- SPG32: Paraplegia with Mental retardation & Brainstem malformation
63
● Chromosome 14q12-q21; Recessive- Epidemiology: 1 Portuguese family
- Onset age: 6 to 8 years
- Clinical
- Pyramidal signs
- Gait disorder: Walking with cane
- Tendon reflexes: Brisk in legs
- Plantar response: Extensor
- Bladder: Normal
- Skeletal: Pes cavus
- Mental retardation: Mild; IQ 50 to 60
- Polyneuropathy: 1 patient
- Ataxia: None
- Progression: Very slow
- Pyramidal signs
- MRI
- Cerebellar & Cortical atrophy
- Pontine dysraphism
- SPG35
68
● Fatty acid 2-hydroxylase (FA2H); Chromosome 16q23.1; Recessive
- Epidemiology: Omani and Pakistani families
- Genetics
- Mutations: Missense & Nonsense; Arg235Cys, p.Arg53_Ile58del
- Some patients: Uniparental chromosome 16 disomy
- Allelic disorder: Leukodystrophy with Spasticity & Dystonia
- FA2H protein
- Membrane bound
- Catalyzes 2-hydroxylation of galactosylceramide & sulfatide
- First step in incorporation of α-hydroxylated GalCer into myelin
- CNS location: Oligodendrocytes
- Required for long-term maintenance of myelin
- Mutations cause
- Reduced FA2H enzyme activity
- Abnormal hydroxylation of myelin galactocerebroside lipid components
- Clinical: NBIA syndrome
- Onset
- Age: 4 to 11 years
- Clinical: Gait disorder, spastic & Foot drop
- Pyramidal signs
- Gait disorder: Progression to wheelchair
- Spasticity: Legs > Arms
- Tendon reflexes: Brisk in legs
- Plantar response: Extensor
- Dysarthria: Mild
- Bladder: Urinary frequency
- Weakness: Legs
- Other CNS
- Cognition: Mental retardation (2 patients); Poor school performance
- Seizures: 2 patients
- Dystonia
- Sensation: Normal
- Course: Progressive
- Onset
- Laboratory
- MRI: Unremarkable or Leukodystrophy
- Bilateral & symmetric T2 hyperintensity in cerebral white matter
- Atrophy: Cerebellum, Brainstem, Cervical spinal cord
- Nerve conduction studies: Motor & Sensory normal
- Muscle biopsy: Denervation & Reinnervation
- MRI: Unremarkable or Leukodystrophy
- Spastic paraplegia with Leukodystrophy & Dystonia
72
● Fatty Acid 2-Hydroxylase (FA2H); Chromosome 16q23.1; Recessive
- Epidemiology: Consanguinous Arab-Muslim families; 9 patients
- Genetics:
- Mutations
- Intronic: Homozygous, c.786+1G/A
- Missense: Homozygous, D35Y
- Allelic disorder: SPG 35
- Mutations
- FA2H protein
- Membrane bound
- Catalyzes 2-hydroxylation of galactosylceramide & sulfatide
- Clinical: NBIA syndrome
- Onset
- Age: Childhood; 4 to 6 years
- Gait disorder
- Spasticity: Legs
- Dystonia: Trunk, Limbs & Face; Dysarthria
- Cerebellum: Dysmetria; Dysdiadochokinesis
- Cognitive dysfunction: Some patients
- Seizures: Mild; Some patients
- Progression: Rapid or Slow; Some lose ambulation in teens
- Onset
- Laboratory
- VEPs: Delayed
- EMG & NCV: Normal
- Brain MRI: Leukodystrophy
- Early: Abnormal periventricular white matter
- Tract involvement: Posterior limbs of internal capsules; Corticospinal
- Thinning of corpus callosum & pons
- SPG43
75
● c19orf12; Chromosome 19q12; Recessive
- Epidemiology: 1 Mali (Bambra) family
- Genetics
- Mutation: Ala63Pro
- Allelic disorder: NBIA4 (MPAN)
- c19orf12 protein
- Clinical
- Onset age: 7 to 12 years
- Pyramidal signs
- Gait disorder: Spastic
- Tendon reflexes: Brisk at knees; Reduced or increased at ankles
- Plantar response: Extensor
- Bulbar: Dysarthria in 1 patient
- Bladder: Normal
- Motor neuronopathy: Weakness & Wasting
- Hands & Feet
- Proximal leg weakness
- Contractures: Knees & Ankles
- Cognition: Normal
- Ataxia: None
- Progression: Slow; Ambulation present
- Same mutation may produce
270
- Optic atrophy
- Motor neuronopathy
- No spasticity
- SPG45: Spastic Paraplegia with early onset (SPG65)
74,
97
● 5'-Nucleotidase, cytosolic II (NT5C2);
Chromosome 10q24.32; Recessive
- Epidemiology: 10 patients
- Genetics
- Mutations
- Splice: c.175+1G>A; c.988-1G>T
- Frameshift: c.1225delA
- Stop: c.G86A (R29X); c.A445T (R149X)
- Mutations
- NT5C2 protein function
- Preferentially hydrolyzes IMP
- Purine & Pyrimidine nucleotide metabolism
- Clinical: Uncomplicated or Complicated SPG
- Onset age: Infancy to 18 months
- Upper motor neuron
- Spasticity: Mild; Legs > Arms
- Tendon reflexes: Increased
- Gait: Unsteady
- Strength: Normal
- Intellectual disability
- Ocular: Optic atrophy; Myopia; Some patients
- Skeletal: Some patients
- Contractures: Toes, Knees
- Hyperextension: Knees
- Pes equinovarus
- Laboratory
- MRI: Corpus callosum thin
- SPG46: Spastic Paraplegia with Mental impairment & Thin Corpus callosum
76
● β-Glucosidase 2 (Non-lysosomal glucosylceramidase; GBA2); Chromosome 9p13.3; Recessive
- Epidemiology: Tunisian & Italian families
- Genetics
- Mutations: Trp173*, Arg234*, Arg630Trp, c.1471_1474dupGGCA; Arg879Gln; Arg399* (Recurrent)
- Allelic with: Spastic Ataxia
- GBA2 protein
- Clinical
- Onset ages: 2 to 10 years
- Spastic paraplegia
- Legs > Arms
- Spasticity
- Tendon reflexes: Brisk
- Plantar reflexes: Extensor
- Bladder dysfunction (50%)
- CNS
- Mental retardation
- Cerebellar signs: Mild
- Pseudobulbar dysarthria
- Strength: Normal
- Skeletal: Pes cavus, Scoliosis
- Cataracts: Congenital
- Polyneuropathy (27%): Axon loss
- Disease progression: Slow
- Hypogonadism
- Laboratory
- EMG & NCV: Normal
- MRI
- Thin corpus callosum
- Cerebellar & Cerebral atrophy
- Variant syndrome: Spastic Ataxia
92
● β-Glucosidase 2 (Non-lysosomal glucosylceramidase; GBA2); Chromosome 9p13.3; Recessive
- Epidemiology: Tunisian & Cypriot families
- Genetics
- Mutations: Nonsense & Missense; Tyr121*, Arg340*, Arg873His
- Allelic with: SPG 46
- GBA1 mutations cause Gaucher disease
- GBA2 protein
- Cell membrane
- Catalyzes conversion of glucosylceramide to free glucose & ceramide
- Involved in sphingomyelin generation
- Clinical
- Onset
- Age: Childhood to Teens; 3 to 15 years
- Gait ataxia
- Ataxia
- Gait: Wide based
- Dysarthria
- Truncal
- Eyes: Saccadic pursuit; Nystagmus
- Upper motor neuron features
- Legs > Arms
- Tendon reflexes: Brisk in legs
- Spasticity: Legs > Arms
- Plantar response: Extensor
- Bladder dysfunction
- Peripheral nerve: Position sense loss in some patients
- Skeletal (50%): Pes cavus; Scoliosis
- Hearing loss
- Disease course: Slow progression; Wheelchair after 2 to 3 decades
- Other, occasional
- Ophthalmoparesis (Jerky pursuit & Slow saccades)
- Intellectual disability (Mild)
- Sphincter disorders
- Head tremor
- Onset
- Laboratory
- NCV: Axonal neuropathy; Sensory ± Motor
- EMG: Denervation
- MRI: Head & Spine normal or Cerebellar atrophy
- SPG47: Spastic Paraplegia with Thin Corpus callosum
84,
130
● Adaptor-related protein complex 4, Beta-1 subunit (AP4B1)
Chromosome 1p13.2; Recessive
- Epidemiology: Multiple families
- Genetics
- Mutations: Stop
- Allelic with: CPSQ5
- AP4B1 protein
- Clinical
- Onset age: Infancy
- Spastic paraparesis
- Gait: Spastic; Loss of ambulation
- Tendon reflexes: Brisk
- Plantar reflexes: Extensor
- Course: Slowly progressive
- Mental retardation: Delayed psychomotor development
- Seizures
- Head: Face dysmorphism; Microcephaly
- Laboratory
- Brain MRI
- Corpus callosum thin
- Periventricular white matter abnormalities
- Ventriculomegaly: Asymmetric
- Brain MRI
- SPG48: Spastic Paraplegia
77
● Adaptor-related protein complex 5, Zeta-1 subunit (AP5Z1; KIAA0415)
;
Chromosome 7p22.1; Recessive
- Epidemiology: 5 families
- Genetics: Mutations
- Complex indel in exon 2: c.[80_83del4;79_84ins22]
- Insertion is imperfect quadruplication of CTGTAA(A)
- Generates frameshift & stop codon following amino-acid 29 (p.R27LfsX3)
- Other: Stop & Missense
- AP5Z1 protein
- Clinical
- Onset ages: 13 to 60 years
- Spastic paraparesis
- Gait disorder
- Urinary incontinence
- Polyneuropathy: Sensory-Motor
- CNS: Ataxia; Dystonia; Parkinsonism
- Progression: Slow
- Laboratory
- Brain MRI: Periventricular white matter hyperintensities
- Spine MRI: Cord hyperintensities in 1 patient
- Retina: Depigmented zones
- Skin fibroblasts: Autofluorescent & lamellar lysosomal storage material
- SPG49: Spastic Paraplegia, Complex, with Thin Corpus Callosum
90
● Tectonin beta-propeller repeat containing 2 (TECPR2)
;
Chromosome 14q32.31; Recessive
- Nosology: HUGO designates SPG49 as being due to CYPUI mutations (SPG56)
- Epidemiology: Jewish Bukharian families
- Genetics
- Mutation: 1 bp frameshift; Leads to premature stop; c.3416delT
- Allelic disorder: Dysautonomia with Intellectual disability (HSAN9)
- TECPR2 protein
156
- Autophagy: Positive regulator
- Interacts with six human Atg8 homologs, including LC3
- Effector of early endosomal Rab protein, Rab5
- Membrane recruitment
- Recycling of Cargo receptors: α5β1 integrins → Lysosomal degradation
- Interacts with: SNX17 & subunits of WASH complex
- Regulate formation of actin-dependent cargo retrieval subdomain on early endosomes
- Clinical
- Onset
- Age: < 2 years
- Hypotonia
- Developmental delay
- Spastic paraparesis
- Gait: Spastic, Rigid, Ataxic
- Speech: Dysarthric
- Intellectual disability: Severe
- Face: Hypomimic
- Respiratory disorders
- Central hypoventilation, episodic
- Wake apnea: With disease progression
- Dysmetria: Arms
- Tendon reflexes: Absent
- Seizures: Occasional patient
- Gastroesophageal reflux: Recurrent pulmonary infections
- Skeletal: Dysmorphic features
- Short stature, mild
- Brachycephalic microcephaly,
- Face: Round; Low anterior hairline, Dental crowding
- Neck: Short broad
- Chubby appearance
- Onset
- Laboratory
- Metabolic testing: Normal
- MRI
- Corpus callosum: Thin
- Atrophy: Cerebral & Cerebellar (Vermis)
- Muscle biopsy: Normal
- EMG & Nerve conduction: Normal
- EEG: Diffuse slowing
- TECPR2 variant syndrome: Dysautonomia with Intellectual disability (HSAN9; HSAN IX)
105
- Epidemiology
- > 10 families
- Pathogenic carrier frequencies
- General 1:1221
- Ashkenazi-Jewish 1:155
- South Asian 1:7654
- Genetics
- Mutations
- Types: Stop or Missense
- Missense: Nü& Cüterminal regions containing β-propeller repeats
- Ashkenazi-Jewish founder mutation: c.1319delT
- Inheritance: Recessive
- Mutations
- Clinical
- Onset age: Congenital or 1st year
- CNS
- Intellectual disability
- Delayed global psychomotor development
- Speech delay
- Hyperactivity (30%)
- Walking: Very delayed
- Sleep disorder: Apnea, central
- Ataxia: Gait
- Vision impairment (40%)
- Intellectual disability
- Hypotonia
- Dysarthria
- Autonomic
- GI: Vomiting; GE reflux; Dysphagia
- Cough
- Blood pressure: Instability
- Other: Temperature instability; Hyperhidrosis
- Respiratory
- Apnea, central; Hypoventilation; Infections
- Common cause of morbidity
- Polyneuropathy
- Sensory loss: Pain (25%); Ataxia in some
- Tendon reflexes: Reduced in legs
- Skeletal
- High arched palate
- Pectus carinatum
- Short stature & neck
- Face: Dysmorphism
- Plantar response: Extensor
- Laboratory
- Sympathetic skin response: Autonomic neuropathy
- EMG: Normal
- NCV: Normal
- EEG: Normal in most
- Nerve biopsy: Normal
- Brain MRI: Cerebral & Vermian atrophy; Corpus callosum thin (50%)
- Epidemiology
- SPG53: Spastic Paraplegia with Mental Impairment
● Vacuolar protein sorting 37, yeast, homolog of, A (VPS37A; HCRP1);
Chromosome 8p22; Recessive
- Epidemiology: 2 Arab Moslem families, 9 patients
- Mutation: Lys382Asn; Missense; Homozygous
- VPS37A protein
- Location: LAMP1-positive endosomes
- Subunit of the endosomal sorting complex
- Required for transport I (ESCRT-I) complex
- May play a role in ubiquitination
- Clinical
- Onset age: Early childhood; 7 to 36 months
- Delay
- Developmental: Cognitive & Speech
- Motor: Difficulty standing & walking
- Spasticity
- Legs > Arms
- Tendon reflexes: Increased
- Clonus
- Vibration sense: Reduced in some
- Skeletal
- Kyphosis
- Pectus carinatum
- Hyperflexibility: Small joints
- Hypertrichosis (30%)
- Laboratory
- Brain MRI: Usually normal; Occasional white or gray matter changes
- Normal: EMG, Muscle biopsy & EEG
- SPG54: Spastic Paraplegia with Thin Corpus Callosum
89
● Phospholipase DDHD2
;
Chromosome 8p11.23; Recessive
- Epidemiology: > 20 families
- Mutations
- > 35 described
- Location: DDHD domain & other
- Often private
- Stop, Frameshift. or Missense
- Allelic disorder: Spastic-Ataxia, Adult onset
- Phospholipase DDHD2 protein
- Intracellular phospholipase A1 (iPLA1)
- Localization: cis-Golgi; ER -Golgi intermediate compartment
- Hydrolyzes preferentially phosphatidic acid & phosphatidylethanolamine.
- May be involved maintenance of endoplasmic reticulum or Golgi
- Synaptic function: Active zones; Facilitates membrane & vesicle fusion
- Organelle biogenesis
- Membrane trafficking
- Clinical
- Onset age: < 2 years
- Spastic paraplegia
- Legs & Arms
- Gait: Spastic
- Tendon reflexes: Brisk
- Urinary or fecal incontinence (33%)
- Sensation: Normal
- Intellectual disability
- Eye: Strabismus; Optic nerve hypoplasia
- Contractures: Foot
- Laboratory
- MRI
- Thin corpus callosum
- Periventricular white-matter hyperintensities
- Syrinx (50%)
- Mitochondrial function: Normal
- MRS: Lipid accumulation in thalami & basal ganglia
- MRI
- DDHD2 variant: Spastic ataxia syndrome
140
- Epidemiology: 7 families; 1% to 2% of AR SP
- Genetics
- Inheritance: Recessive
- Mutations: Missense or Stop; R112Q, D660H
- Clinical
- Onset age: 3rd to 6th decade
- Spasticity
- Tendon Reflexes: Increased
- Sensory loss: large fiber modalities
- Cognitive Δ: Some patients
- Ataxia (50%)
- Sensory neuropathy
- Laboratory
- Lipid droplets & triglycerides in brain & spinal cord
- SPG56: Spastic Paraplegia with Thin Corpus Callosum
91
● Cytochrome P450, Family 2, Subfamily U, Polypeptide 1 (CYP2U1);
Chromosome 4q25; Recessive
- Nosology: Called SPG49 by HUGO
- Epidemiology: 16 patients
- Mutations: Missense or Deletion; Homozygous or Heterozygous; Asp316Val
- CYP2U1 protein
- Location: Endoplasmic reticulum & Mitochondrial
- ω- and ω-1 fatty-acid (C16¢C22) hydroxylation
- Catalyzes hydroxylation of arachidonic acid & related long-chain fatty acids
- Eicosapentaenoic (EPA) & Docosahexaenoic (DHA) acids
- Metabolites: 19- and 20-hydroxyeicosatetraenoic (HETE) acids
- Clinical
- Onset
- Age: Birth to 8 years
- Gait disorder
- Spastic paraplegia
- Spastic gait
- Tendon reflexes: Increased in Legs > Arms
- Dysarthria: Mild in few patients
- Course: Slow progression
- Dystonia (25%)
- Cognitive delay (20%): May be progressive
- Eye: Pigmentary degenerative maculopathy
- Onset
- Laboratory
- MRI
- May be Normal
- White matter: Lesions & Corpus callosum thin
- Calcifications: Globus pallidus
- NCV: Axonal neuropathy
- Sub-clinical
- Motor-Sensory
- Legs > Arms
- MRI
- SPG59: Spastic Paraplegia
97
● Ubiquitin-specific protease 8 (USP8);
Chromosome 15q21.2; Recessive
- Epidemiology: 1 family, 3 patients
- Genetics: Q310K; Missense mutation
- Clinical
- Onset age: 20 months
- Spasticity
- Gait, Scissors: Walk with support
- Tendon reflexes: Increased in legs
- Progression: Slow
- Sphincter: Normal
- Sensation: Normal
- Cognition: Normal or Borderline
- Laboratory
- EMG/NCV: Normal
- Brain MRI: Normal
- SPG60: Spastic Paraplegia
97
● WD repeat domain 48 (WDR48);
Chromosome 3p22.2; Recessive
- Epidemiology: 1 patient
- Genetics
- Mutation: del628E
- WDR48 protein function
- Regulator of deubiquitination
- Clinical: Complicated SPG
- Onset age: 1 year
- Motor: Gait difficulty
- UMN: Increased tone; Knee DTRs increased
- Neuropathy: Ankle DTRs reduced
- Other: Nystagmus; Mild learning disability
- Laboratory
- NCV: Polyneuropathy
- SPG61: Spastic Paraplegia
97,148
● ADP-ribosylation factor-like 6 interacting protein 1 (ARL6IP1);
Chromosome 16p12.3; Recessive
- Epidemiology: 11 patients
- Genetics
- Mutations: Stop > Missense; Deletion
- Biallelic null variants: Severe, early onset disease
- ARL6IP1 protein function
- Clinical: Complicated SPG
- Onset age: Prenatal to 14 months
- Prenatal: Severe, early onset group
- Reduced movements
- Growth retardation
- Arthrogryposis, Neurogenic
- Development
- Hypotonia: Axial
- Motor delay
- Face: Dysmorphism
- Respiratory insufficiency
- Regression in some
- Pyramidal (UMN): Spasticity
- Scissor gait
- Knee DTRs brisk
- Gait: Unsteady or Non-ambulatory
- Tendon reflexes
- Ankles: Absent
- Other: Brisk or Absent
- Plantar response: Extensor
- Polyneuropathy
- Sensory loss
- Acromutilation: Asymptomatic bone fractures
- Tongue: Fasciculations in early onset patient
- Muscle pathology: Denervation
- Cognition: Normal
- Course: Death in childhood in some
- Laboratory
- NCV: Motor & Sensory axon loss, Legs > Arms
- Muscle: Denervation
- Brain MRI: Cortical development malformations
- Subcortical atrophy
- Leukoencephalopathy
- Corpus callosum: Agenesis
- Serum CK: Normal
- SPG62: Spastic Paraplegia
97
● ER lipid raft associated 1 (ERLIN1);
Chromosome 10q24.31; Recessive
- Epidemiology: > 20 patients
- Genetics
- Mutations
- Stop: c.C763T; p. R255X
- Missense: c.G149T ;p.G50V
- Frameshift: c.862_868delACCAGG; p.del203-204YQ
- Allelic disorder
- Mutations
- ERLIN1 protein function
- ER-associated degradation
- Lipid rasft protein
- Subcellular: Endoplasmic reticulum; Nuclear envelope
- Homologous protein: ERLIN2 (SPG18)
- ER disorders
- Clinical
- Onset age: 17 months to 13 years
- UMN
- Spasticity: Legs > Arms
- Gait: Unsteady; spastic
- DTRs: Increased in most
- Plantar reflex: Extensor
- Ataxia (50%)
- Amyotrophy (50%)
- Skeletal
- Contractures: Knees (20%)
- Scoliosis
- Intelligence: Borderline or Normal
- Laboratory
- EMG/NCV: Normal
- Brain MRI: Normal
- ERLIN1 variant syndrome: ALS, Early onset, Slow progression
119
- Epidemiology: Turkish family
- Genetics
- Inheritance: Recessive
- Mutation: Homozygous; Missense; Val94Ala
- Clinical
- Onset age: 2nd decade
- Spasticity
- Gait: Onset in teens
- Bulbar: Onset in 5th decade
- Plantar response: Extensor
- Tendon reflexes: Brisk
- Lower motor neuron: Hand atrophy
- Course: Slow progression over decades
- Laboratory
- EMG: Denervation in tongue, arms & legs
- SPG63: Spastic Paraplegia + Thin corpus callosum
97
● Adenosine monophosphate deaminase 2 (AMPD2);
Chromosome 1p13; Recessive
- Epidemiology: 2 patients
- Genetics
- Mutation: c.318delT; p.C107Afs365X; Frameshift
- Allelic with: Pontocerebellar hypoplasia 9 (PCH9)
- AMPD2 protein function
- Deaminates AMP to IMP in purine nucleotide metabolism
- Maintenance of cellular guanine nucleotide pools
- Regulates feedback inhibition of adenosine derivatives on de novo purine synthesis
- Expression: Widespread; Type I & Smooth muscle; Nerve
- Also see: AMPD1
- Clinical: Complicated
- Onset age: 1 to 2 years
- UMN
- Delayed walking
- Gait: Scissors
- Tone: Increased
- DTRs: Increased
- Plantar response: Extensor
- Cognition: Normal
- Sensation: Normal or Reduced
- Laboratory
- MRI: Periventricular WM changes; Corpus callosum thin
- AMPD2 Variant syndrome: Pontocerebellar hypoplasia 9 (PCH9)
- AMPD2 Genetics
- Inheritance: Recessive
- Mutations
- Null alleles or Missense: Some have residual activty
- D552fs; E778D; Y349X; R674H; D793Y
- Allelic with: SPG63
- AMPD2 protein
- Clinical
- Microcephaly
- Hypotonia
- Spasticity
- Limbs
- Tendon reflexes: Increased
- Contractures: Progressive
- Seizures: Some patients
- Face: Dysmorphic features
- Course
- Laboratory
- MRI
- Cerebellum & Pons: Size reduced ≥ 50%
- Posterior fossa: Mega cisterna magna; Flat ventral pons (Fluid-filled)
- Atrophy: Cerebral cortex
- Corpus callosum: Hypoplasia or Aplasia
- EEG: Epileptic activity
- NCV: Neuropathy, Axonal
- MRI
- AMPD2 Genetics
- SPG64: Spastic Paraplegia
97
● Ectonucleoside triphosphate diphosphohydrolase 1 (ENTPD1);
Chromosome 10q24.1; Recessive
- Epidemiology: 2 families, 4 patients
- Genetics
- Mutations
- Missense: c.G649A; p.G217R
- Stop: c.G719T; p.E181X
- Mutations
- ENTPD1 protein function
- Hydrolyzes ATP & other nucleotides
- Regulates purinergic transmission
- Clinical: Complicated SPG
- Onset age: 1 to 4 years
- UMN: Spasticity; Abnormal gait; Plantar reflex extensor
- CNS, other
- Intelligence: Moderate disability or Borderline
- Aggressive behavior
- Amyotrophy (75%)
- DTRs: Varied
- Puberty: Delayed
- Some patients
- Microcephaly
- Cataract, congenital
- Pes equinovarus
- Laboratory
- Brain MRI: White matter changes, mild
- SPG66: Spastic Paraplegia
97
● Arylsulfatase family, member I (ARSI);
Chromosome 5q32; Recessive
- Epidemiology: 1 patient
- Genetics
- Mutation: c.1641insA; p.C548Mfs559X; Frameshift
- ARSI protein function
- Hydrolyze sulfate esters
- Hormone biosynthesis
- Clinical: Complicated SPG
- Onset age: 1.5 years
- Spasticity
- Tendon reflexes: Absent
- Gait: Non-ambulatory
- Sensory loss
- Intelligence: Borderline
- Contractures: Pes equinovarus
- Laboratory
- NCV: Sensory-Motor neuropathy
- MRI: Corpus callosum & Cerebellar hypoplasia, Colpocephaly
- SPG67: Spastic Paraplegia
97
● Post-GPI attachment to proteins 1 (PGAP1);
Chromosome 2q33.1; Recessive
- Nosology: Neurodevelopmental disorder + Dysmorphic features, Spasticity, Brain abnormalities (NEDDSBA)
- Epidemiology: 5 families
- Genetics
- Mutation: c.1952+1G>T; Splice
- PGAP1 protein
- Endoplasmic reticulum
- GPI biosynthesis
- GPI inositol deacylase
- Catalyzes inositol deacylation of glycosylphosphatidylinositol (GPI)
- ER-to-Golgi transport of GPI-anchor proteins
- Clinical
- Onset age: 4 months to 4 years
- Motor delay
- Cognition: Borderline to normal
- Spasticity
- Tendon reflexes: Increased in legs
- Tremor
- MRI: Variable
- Corpus callosum agenesis
- Cortical atrophy
- Vermis atrophy
- Nosology: Neurodevelopmental disorder + Dysmorphic features, Spasticity, Brain abnormalities (NEDDSBA)
- SPG69: Spastic Paraplegia
97
● RAB3 GTPase activating protein subunit 2 (RAB3GAP2);
Chromosome 1q41; Recessive
- Epidemiology: 1 patient
- Genetics
- Mutation: c.T1955G; p.L252X; Stop codon
- Allelic disorders
- Martsolf syndrome 1
- Warburg micro syndrome 2
- Muscular dystrophy/Short stature
- Martsolf syndrome 1
- RAB3GAP2 protein function
- Exocytosis of neurotransmitters & hormones
- Autophagy: Positive modulator
- GTPase activating complex subunit: Specificity for RAB3 Rab-GTPase activating proteins
- Clinical: Complicated SPG
- Onset age: 6 months
- Developmental delay: Global
- Spasticity
- Dysarthria
- Tendon reflexes: increased in legs
- Plantar reflex: Extensor
- Deafness
- Cataracts
- Cognition: Intellectual disability
- Brain MRI: Normal
- RAB3GAP2 variant: Muscular dystrophy/Short stature
134
- Epidemiology: Mennonite sibling pair
- Genetics
- Inheritance: Recessive
- Mutation: Missense; Homozygous; Arg420Cys
- Clinical
- Onset age: 2nd decade
- Skeletal: Microcephaly; Short stature
- Tracheomalacia
- Face: Inexpressive; Eyebrows arched; Philtrum long; Upper lip thin
- Eyes: Ptosis
- Weakness: Proximal; Arms, Scapulae & Legs; Dysphagia
- Muscle mass; Reduced
- Laboratory
- EMG: Myopathic
- Serum CK: 3,200
- SPG71: Spastic Paraplegia
97
● Zinc finger RNA binding protein (ZFR)
;
Chromosome 5p13.3; Recessive
- Epidemiology: 1 patient
- Genetics
- Mutation: c.T956C; p.L319P; Missense
- ZFR protein function
- ? RNA localization
- Clinical: Uncomplicated SPG
- Onset age: 1 year
- Spasticity
- Gait: Scissors or Lost
- Tendon reflexes: Increased in legs
- Plantar reflex: Extensor
- Cognition: Normal
- Laboratory
- MRI: Corpus callosum thin
- EMG/NCV: Normal
- SPG75: Spastic Ataxia 75
97
● Myelin Associated Glycoprotein (MAG);
Chromosome 19q13.12; Recessive
- Epidemiology: 12 patients
- Genetics
- Mutations: Missense (Cys430Gly, Ser133Arg) & Nonsense
- Allelic disorders
- Pelizaeus-Merzbacher-like
- Cerebellar Ataxia + Oculomotor Apraxia
- MAG protein
- Other disorder: MAG neuropathy
- Clinical
- Onset age: 1 to 3 years
- Developmental delay
- Spasticity: Gait; Tendon reflexes increased
- Ataxia: Gait; Dysarthria; Dysmetria; Tremor
- Amyotrophy
- Sensory loss: Legs
- Tendon reflexes: Reduced
- Dystonia
- Intellectual disability
- Eyes: Nystagmus; Optic atrophy; Oculomotor Apraxia in 1 family
- Laboratory
- Brain MRI: Optic nerve, Cerebellum & Corpus callosum atrophy
- NCV: Axon loss
- MAG variant: Pelizaeus-Merzbacher-like (PML) Infant-onset disorder
- Epidemiology: 5 families; 7 patients
- Genetics
- Mutations: Missense (c.399C>G, p.S133R; Homozygous); Null
- Inheritance: Recessive
- Clinical
- Onset age; Infancy
- Mental retardation
- Spastic paraparesis: Legs > Arms; Dysarthria
- Tendon reflexes: Reduced
- Ataxia
- Eye: Nystagmus; Optic atrophy
- Extrapyramidal
- Course: Slow progression
- Laboratory
- NCV
- Distal motor latencies: Prolonged
- Velocity: Reduced
- Brain MRI
- Corpus callosum & Cerebelum atrophy
- White matter hyperintensities
- Nerve pathology
- MAG: Absent
- Onion-bulbs: Ill-formed
- Myelin sheath: Thin
- NCV
- SPG76: Spastic Paraplegia 76
109
● Calpain 1 (CAPN1);
Chromosome 11q13.1; Recessive
- Nosology: Spastic ataxia
- Epidemiology: 35 families
- Genetics
- Mutation types: Nonsense, Missense, Frameshift, Splice-site
- Mutation locations: Functional domains of CAPN1 protein; No hotspot
- Allelic disorder: Spinocerebellar ataxia in Parson Russell Terrier dogs
- CAPN1 protein
127
- Location: Central nervous system; Muscle
- Heterodimer: With small subunit CAPNS1
- Protease
- Calcium-dependent
- Neutral
- Non-lysosomal: Not degredative
- Cleaves vesicle fusion protein dysferlin
- Role in: Repair of mechanical membrane damage
- Overlap with Calpain-2
- Loss of both CAPN1 & CAPN2
by knockout of CAPNS1
: Myopathy
- No effect of calpains on recovery from membrane perforation by streptolysin-O or saponin
- Clinical: Complicated or Pure HSP
- Onset age: 19 to 38 years; Mean 28 years
- Spasticity
- Legs > Arms
- Gait
- Hyperreflexia: Legs ± Arms
- Extensor plantar response (80%)
- Weakness (60%): Legs ± Arms; Distal
- Dysarthria (80%)
- Bladder dysfunction (20%)
- Foot deformity (60%): Pes cavus
- Ataxia (70%): Limb; Gait
- Sensory: May be normal
- Polyneuropathy (20%): Sensory loss
- Ocular: Saccades slow in 1 family
- Course
- Progressive
- Females: More likely to develop complicated HSP
- Laboratory
- NCV: Normal or Axonal sensory neuropathy
- MRI: Normal or Cervical spinal cord atrophy or Cerebellar atrophy
- CAPN1 variant syndrome: Spinocerebellar ataxia in Parson Russell Terrier dogs
- SPG83: Spastic Paraplegia
● 4-Hydroxyphenylpyruvate Dioxygenase-like (HPDL);
Chromosome 1p34.1; Recessive
- Epidemiology: 15 families
- Genetics
- Mutations: Gly50Asp
- Allelic disorder: NEDSWMA
more with Truncating mutations
- HPDL protein
- Mitochondrial
- Oxygenase: Vicinal oxygen chelate (VOC) superfamily
- Metalloenzyme: Iron-containing, non-heme
- Clinical
- Onset age: Mid-Teens
- Gait: Unsteady
- Muscle: Cramps; Myalgia, Poor exercise tolerance
- Spastic paraparesis: Leg DTRs increased; Plantar response extensor; Bladder urgency
- Course: Progressive; May lose walking
- Other: Dysarthria, Dysphagia
- Cognition: Normal
- Laboratory
- Brain & Spine MRI: Normal
- SPG84: Spastic Paraplegia
● Phosphatidylinositol (PI) 4-kinase, Catalytic α (PI4Ka);
Chromosome 22q11.21; Recessive
- Epidemiology: 2 patients
- Genetics
- Mutations: Hypomorphic; Deletion & Missense
- Allelic disorder
- Polymicrogyria, perisylvian + Cerebellar hypoplasia & Arthrogryposis (PMGYCHA)
- Polymicrogyria, perisylvian + Cerebellar hypoplasia & Arthrogryposis (PMGYCHA)
- PI4Ka protein
- Catalyzes 1st step in biosynthesis of Phosphatidylinositol 4,5-bisphosphate
- Clinical
- Onset age: Childhood to Teens
- Spastic paraparesis
- Legs
- Tendon reflexes: Increased
- Hand weakness
- Pes cavus
- Nystagmus
- Cognitive: Impairment with younger onset
- Infections, Recurrent
- GI: inflammatory bowel disease; Multiple intestinal atresia
- Laboratory
- Brain MRI: Cervical spinal cord atrophy
- Immune: B-Cell Lymphopenia & Hypogammaglobulinemia 147
- SPG86: Spastic Paraplegia
● ABHydrolase Domain-containing Protein 16A, phospholipase (ABHD16A);
Chromosome 6p21.33; Recessive
- Epidemiology: 9 families; 18 patients
- Genetics
- Mutations: Missense & Stop
- ABHD16A protein
- Endoplasmic reticulum membrane protein
- Main brain phosphatidylserine (PS) hydrolase
- Colocalizes with ABHD12
- General
- Lipid metabolism
- Intracellular signaling
- Nosology
- Phosphatidylserine lipase ABHD16A
- α/β Hydrolase domain-containing protein 16A
- Clinical
- Onset age: Infancy or Early childhood
- Spasticity
- Seizures: Some patients
- PNS: Axon loss in some patients
- Developmental delay: Global
- Face: Dysmorphic
- Skeletal: Scoliosis; contractures
- Skin pigmentation: Increased or Decreased
- Course: Progressive
- Laboratory
- Brain imaging: White matter & corpus callosum Δ;
- SPG87: Spastic Paraplegia
● Transmembrane protein 63C (TMEM63C);
Chromosome 14q24.3; Recessive
- Epidemiology: 3 families, 7 patients
- Genetics
- Mutations: Deletion, Duplication, Stop
- TMEM63C protein
- Channel subunit: Hyperosmolarity-activated ion currents
- ER & Mitochondrial morphologies & dynamics
- Clinical
- Onset: Infancy to 19 mo
- Spasticity: Paraparesis; DTRs brisk
- Spinal: Lumbar lourdosis
- Other CNS: Intellectual disability; Speech delay
- Laboratory
- Nerve conductions & EMG: Normal
- Brain MRI: Normal
- SPG88: Spastic Paraplegia
● Karyopherin α-3 (KPNA3);
Chromosome 13q14.2; Dominant
- Epidemiology: 6 families; 11 patients
- Genetics
- Mutations: Missense; Often Leu
- KPNA3 protein
- Subunit of a nuclear transport receptor (NTR)
- Nucleocytoplasmic trafficking
- Transport HNRNPK
& PCAF (KAT2B)
- Clinical
- Onset age: Infant
- Spasticity
- Walking: Delayed or None
- Tendon reflexes: Brisk
- Intellectual disability: Some patients
- Course: Slow progression
- Laboratory
- Brain MRI: White matter Δ; Cerebellar atrophy; Some normal
- SPG89: Spastic Paraplegia, Childhood onset
● Autocrine Motility Factor Receptor (AMFR; GP78);
Chromosome 16q13; Recessive
- Epidemiology: 20 patients, 8 families
- Genetics
- Mutations: Loss of function; Often homozygous
- AMFR protein
- RING-H2 finger E3 ubiquitin ligase
- Anchored at endoplasmic reticulum (ER)
- Mediates polyubiquitination: HMGCR & INSIG1
- ER-associated degradation (ERAD)
- Clinical
- Onset age: Childhood
- Spasticity
- Tendon reflexes: Increased
- Plantar response: Extensor
- Gait: Unsteady
- Paraparesis: Most
- Quadriparesis: Severe patients; May also have seizures
- Muscle cramps
- Motor delay: Late walking
- Developmental delay (50%): Intellectual; Learning disabilities
- Laboratory
- Brain MRI: Corpus callosum thin
- SPG: Spastic paraplegia & Psychomotor retardation ± Seizures (SPPRS)
● HECT Domain- and ankyrin repeat-containing E3 ubiquitin protein ligase 1 (HACE1; KIAA1320);
Chromosome 6q16.3; Recessive
- Epidemiology: 6 families; 14 patients
- Genetics
- Mutations: Often stop
- Allelic disorders
- Lung cancer mutations
- Ataxia, Norwegian elkhound, black
- HACE1 protein
133
- E3 ubiquitin ligase
- Regulates activity of cellular GTPases
- Tumor suppressor
- Mutations: Disordered autophagy, mitophagy & oxidative function
- Clinical
- Onset: Birth
- Psychomotor development: Delayed
- Spasticity: Legs; Gait disorder; Dysarthria; Slow progression
- Seizures (60%): Especially myoclonic
- Ataxia: Gait
- Other: Some patients
- Ocular: Strabismus, Myopia, Retinal dystrophy
- Face: Hypotonia
- Dystonia
- Hearing loss: Sensorineural
- Skeletal: Kyphosis, Scoliosis, Hip dislocation, Foot deformities; Large head
- Genitalia: Hypoplastic in males
- Laboratory
- Brain imaging: Cerebral atrophy; Corpus callosum thin
- SPG 90: Spastic paraplegia, Complex
139
● Serine palmitoyltransferase, small subunit, A (SPTSSA);
Chromosome 14q13.1; Recessive or Dominant de novo
- Epidemiology: 3 patients
- Genetics
- Mutations: Thr51Ile (Dominant); Gln58X (Recessive)
- Allelic disorders
- SPG90A
: Dominant
- SPG90B
: Recessive
- SPG90A
- SPTSSA protein
- Location: Endoplasmic reticulum membrane
- Catalyzes 1st, & Rate-limiting, step in sphingolipid biosynthesis
- Small SPT subunit
- Stimulates SPT activity
- Confers Acyl-CoA preference (C14/C16) to SPT catalytic heterodimer of SPTLC1 + SPTLC2 or SPTLC3
- Role in MBOAT7
location to mitochondria-associated membranes
- SPTSSA Mutants: Impaired ORMDL regulation; Excessive sphingolipid synthesis
- Clinical
- Onset age: Early childhood
- Failure to thrive
- Spastic paraplegia
- Language/Cognitive dysfunction
- Hearing loss: Sensorineural
- Short stature
- GERD
- Skin: Vessel malformation
- Laboratory
- EEG: Spikes; Slowing
- Brain MRI: Volume loss; White matter changes
- SPG 92: Spastic paraplegia ± Mild cognitive Defects
● FIC Domain-containing protein Adenylyltransferase (FICD: HYPE);
Chromosome 12q23.3; Recessive
- Epidemiology: 5 patients, 4 families
- Genetics
- Mutations: Arg374His, 2 bp deletion
- FICD protein
- Endoplasmic reticulum
- Bifunctional enzyme
- AMP transferase: Posttranslationally attaches AMP moiety to target proteins
- Phosphodiesterase: De-AMPylates AMPylated proteins to release AMP moiety
- Regulates activity of BiP (HSPA5/GRP78)
: Chaperone & Regulator of Unfolded protein response (UPR)
- Clinical
- Onset age: 1 to 16 years; Median 5 years
- Spasticity
- Legs > Arms
- Gait disorder
- Bladder dysfunction (60%): Urgency
- Tendon reflexes: Increased in legs
- Plantar response: Extensor
- Sensation: Vibration reduced in legs in some patients (60%)
- Cognitive impairment (60%): Mild; Later onset
- Cerebellar: Saccadic pursuit; Tremor (Postural)
- Course: Slow progression
- Laboratory
- NCV: Motor ± Sensory polyneuropathy; Axon loss
- Brain MRI: Often normal
Spasticity + Systemic Disorders
- Sjögren-Larsson Syndrome (SLS)
17
● Fatty aldehyde dehydrogenase (ALDH3A2; ALDH10; FALDH)
;
Chromosome 17p11.2; Recessive
- Epidemiology
- Common in Northern Sweden
- Other areas 0.4 per 100,000
- Genetics
- ~ 50 different mutations
- Missense & Deletion most common
- FALDH protein
- Carboxyl terminal hydrophobic: Microsome membrane anchor
- Catalyzes medium & long chain fatty aldehydes to carboxylic acids
- Deficiency
- ? Increased fatty alcohols or aldehyde modified molecules
- Abnormal cell membrane integrity
- High levels of biologically active lipids
- Clinical
- Generally homogeneous phenotype
- Onset: Pre-term birth
- Spasticity
- Quadriparesis: Onset < 2 years
- Spastic dysarthria
- Slow progression: Disability; Contractures
- Other CNS
- Mental Retardation: Non-progressive
- Seizures
- Systemic
- Skin
- Ichthyosis
- Pruritis
- Color: Brownish-yellow
- Eye
- Pigmentary Retinal degeneration (Macular dystrophy)
- Reduced visual acuity
- Crystalline deposits in retina
- Photophobia
- Skin
- Severity
- 90% with severe disability
- Most with some walking, but use wheelchair
- 10% with mild syndromes
- Treatment: Leucotriene B4 (LTB4) synthesis inhibition
- Laboratory
- EEG: Slow background activity
- MRI: Retarded myelination; Hyperintense periventricular
- Urine: High LTB4 & 20-OH-LTB4
- Epidemiology
- Ichthyosis, Intellectual Disability & Spastic Quadriplegia (ISQMR)
85
● Elongation of very long chain fatty acids-like 4 (ELOVL4);
Chromosome 6q14.1; Recessive
- Epidemiology: 3 families, 5 patients
- Genetics
- ELOVL4 mutations
- Stop: Tyr26*; Arg216X; Ile230Metfs*22 (c.690del); Homozygous
- Heterozygous mutations cause: Macular degeneration (SGDT3)
- Allelic disorders
- SCA34
- Stargardt disease 3
- ISQMR
- Other elongase disorders: SCA38 - ELOVL5
- ELOVL4 mutations
- ELOVL4 protein
- Very long chain fatty acid synthesis
- Component of fatty acid elongation system
- Mutation: Deficiency of VLCFAs >28 carbons long (30% of brain myelin)
- Clinical
- Onset age: Birth
- Ichthyosis
- Variable
- Especially neck & diaper areas
- More severe with cold
- CNS: Some patients
- Seizures: Myoclonic epilepsy
- Mental retardation
- Spastic quadriplegia
- Head circumference: Small
- Laboratory
- Brain MRI: Delayed myelination; Atrophy
- ELOVL4 variant disorder: SCA34
99
- Nosology: Erythrokeratodermia with Ataxia
- Epidemiology: > 60 patients
- Genetics
- Inheritance: Dominant, Incomplete penetrance
- ELOVL4 mutations
- Often missense: Leu168Phe, Ile171Thr, Trp246Gly
- Deficient Very Long Chain-Saturated FA biosynthesis
- Clinical
- Onset: Infant to Adult
- Skin disorder (30% to 90%)
- Onset age: Infancy
- Papulosquamous erythematous ichthyosiform plaques
- Similar to erythrokeratodermia variabilis (EKVP)
- Tend to be on extremities
- Subside during summer
- Disappear by age 25
- May reappear after age 40: With onset of ataxia
- Erythrokeratodermia: Other (GJB3)
- Ataxia
- Onset age: Mean 51 years; Range = 3rd to 8th decade
- Gait disorder
- Eyes: Nystagmus; Slow pursuit or saccades
- Dysarthria
- Limbs
- Polyneuropathy (50%)
- Tendon reflexes: Reduced (50%)
- Axon loss: Mild
- Autonomic involvement
- Other
- Retinitis pigmentos
- Pyramidal tract signs
- Cognitive Δ
- Course
- Progression: Slow
- Cane after 10 years
- Walker by 7th or 8th decade
- Severity: Mild to Moderate
- Laboratory
- Brain MRI
- Atrophy: Cerebellum, Pons & Cortex
- Hot Cross Bun Sign (30%)
- Brain pathology
- Pons
- Macrophages (PAS+, Lipopigment, autofluorescent; Trilaminar spicules)
- Storage lipid: Accumulates within neuronal lipofuscin
- White matter: Vacuolar; Degenerated oligodendroglia
- Four repeat tau: Aggregation; Tangles
- Pons
- Brain MRI
- SPG: Ichthyotic Keratoderma, Spasticity, Hypomyelination & Dysmorphic features (IKSHD)
● Elongation of Very long chain Fatty acids-like 1 (ELOVL1);
Chromosome 1p34.2; Recessive
- Epidemiology: 1 family, 2 patients
- Genetics
- Mutation: Ser165Phe
- ELOVL1 protein
- Biosynthesis of C26 fatty acids & sphingolipids
- Clinical
- Ichthyotic keratoderma: Epidermal hyperproliferation & Increased keratinization
- Spastic paraparesis
- Eye: Optic atrophy; Nystagmus
- Face: Dysmorphic features
- Laboratory
- Brain MRI: Hypomyelination
- Spastic paraplegia with Evans syndrome
- Spasticity
- Early childhood onset
- Legs predominantly involved
- Evans syndrome
- Hemolytic anemia: Coombs-positive
- Thrombocytopenia: Immune
- Spasticity
- Fitzsimmons Syndrome
- Epidemiology: 2 families; 6 patients
- Genetics
- Possibly X-linked
- Clinical
- CNS
- Spastic Paraplegia: Progressive
- Dysarthria: Nasal speech
- ± Mental Retardation
- Skeletal disorders: Hands & Feet
- Brachydactyly
- Cone shaped epiphyses
- Abnormal metaphyseal-phalangeal pattern
- Palmoplantar hyperkeratosis
- CNS
- Mothers
- Tendon reflexes: Brisk
- Hand tremor, postural
- Learning difficulties, moderate
- Plantar hyperkeratosis
- SPG23: Spastic paraplegia with pigmentary abnormalities (Lison)
115
● Dual serine/threonine & tyrosine protein kinase (DSTYK; RIP5; RIPK5; DUSTYPK); Chromosome 1q32.1; Recessive
- Epidemiology: Arabian, Israeli & Palestinian-Jordanian families
- Genetics
- Mutation: Complex homozygous 3' 4-kb deletion/20-bp insertion
- Allelic with: Congenital urological developmental disorders, Dominant (CAKUT1)
- DSTYK protein
- Membrane-associated
- Protein kinase
- Regulation of cell death
- Epithelium
- Urinary tract development
- Clinical
- Onset
- Early childhood
- Spastic paraparesis
- Skin
- Facial appearance: Prematurely aged
- Scalp hair: White from birth
- Eyebrows and eyelashes: Premature graying
- Skin
- Covered areas: Hypopigmented
- Sun-exposed areas: Patchy, vitiligo-like depigmentation
- Spastic paraparesis
- Weakness: Legs
- Spasticity: Legs
- Tendon reflexes: Brisk in legs
- Plantar reflexes: Extensor
- Disability: Difficulty walking
- CNS, other
- Mental retardation: Mild
- Head circumference: Reduced in some patients
- Scoliosis: Some patients
- Onset
- Laboratory
- NCV: Axonal neuropathy
- Sensory-Motor
- Leg predominant
- Muscle: Type 1 fiber predominance
- MRI: Mild ventricular enlargement in 1 patient
- NCV: Axonal neuropathy
- Paraparesis, Hypermanganesemia, Polycythaemia & Cirrhosis
● Solute carrier family 30, Member 10 (SLC30A10);
Chromosome 1q41; Recessive
- Epidemiology: 1 patient
- Genetics
- Mutations: c.585del, p.Thr196Profs*17
- Allelic with: Hypermanganesemia with dystonia, polycythemia, and cirrhosis (HMDPC)
- SLC30A10 protein: Manganese transporter
- Clinical
- Onset age: 14 years
- Paraparesis: Spastic; Progressive
- Weakness: Distal legs
- Sensory loss: Distal legs
- Tendon reflexes: Increased in arms; Reduced in legs
- Plantar reflex: Extensor
- No extrapyramidal signs
- Laboratory
- Brain (basal ganglia), liver & blood manganese: High
- Brain MRI: High signal in lenticular & dentate nuclei, brainstem & pituitary
- Polycythemia
- Hepatic dysfunction: Micronodular cirrhosis
- Cardiomyopathy: Hypertrophic
Spasticity + PNS
- SPG17: Silver syndrome; Spastic paraplegia with amyotrophy of hands & feet
● BSCL2 gene (Seipin);
Chromosome 11q12.3; Dominant
- Epidemiology: > 10 families
- Genetics
43
- Mutations
- Missense
- Exon 3
- N88S; S90L
- Mutation effects
- Alter N-glycosylation site
- Aggregate formation
- BSCL2 Allelic disorders
- Berardinelli-Seip congenital lipodystrophy, Type 2: Null mutations
- Hereditary distal motor neuropathy, 5C (HMN5C)
- SPG17
- CMT2-like
- Progressive encephalopathy (PELD)
: Truncating mutations; c.985C-T
- Asymptomatic 4%
- Mutations
- Seipin protein
- Integral membrane protein
- Glycosylated
- Subcellular location: Endoplasmic reticulum
- Expression: Neurons in Spinal cord & Frontal lobe cortex
- Function: Lipid Droplets
- Mutations result in
- Accumulation of unfolded protein in endoplasmic reticulum
- Unfolded protein response
- Clinical
- General: Variable phenotype within families
- Onset
- Age: 2 to 40 years
- Amyotrophy
- Some families with early leg spasticity
- Spastic paraparesis
- Especially legs (100%)
- Tendon reflexes: Brisk
- Wasting & Weakness (83%)
- Distal
- Arms & Legs
- Pes cavus (83%)
- Bladder disorder (18%)
- Sensory: Usually normal; Occasional reduced vibration at ankles
- Course: Slow progression
- Silver syndromes: Other
- SPG20: Troyer syndrome
28
● Spartin;
Chromosome 13q13.3; Recessive
- Epidemiology: Kuwait & Old Order Amish families
- Genetics
- Mutations: 1110delA; 2-BP del, 364AT
- Hypermethylation of the SPG20/spartin promoter: Biomarker of colorectal carcinoma
- Spartin protein
- Sequence similarities: Spastin, SNX15, VPS4, Skd1
- Functions
- Associates with Cardiolipin: via Plant-Related Senescence Domain
- Regulates Mitochondrial Ca++ Homeostasis
- Postulated functions: Other
- Intracellular protein trafficking of late endosomal components
- Association with microtubules & tubulin
- Functions in degradation of epidermal growth factor receptor
- Localization: Mitochondria & Cytoplasm
- Mono-ubiquitinated
- Clinical
- Onset
- Age: Early childhood
- Dysarthria
- Late walking: Mean 16 months
- Upper motor neuron
- Spastic paraparesis: Legs > Arms
- Bulbar: Dysarthria (Spastic); Drooling
- Incontinence: Occasional
- Tendon reflexes: Brisk in legs; Arms increased late in course
- Plantar response: Extensor
- Lower motor neuron
- Weakness: Distal in hands & feet
- Wasting: Distal
- Sensory: Normal
- Cerebellar: Mild signs
- Dysdiadochokinesia
- Terminal dysmetria
- No nystagmus
- Extrapyramidal: Advanced cases
- Dystonic limb posturing
- Choreoathetoid movements
- Cortical
- Developmental delay: Mild
- Emotional lability & Affective disorders
- IQ reduced
- Skeletal
- Short stature
- Contractures: Legs; DIP joints
- Hyperextensible joints: PIP; Wrist
- Progression
- Slow
- Difficulty walking in 4th & 5th decades
- Onset
- MRI: White matter abmormalities
- Temporoparietal periventricular
- Posterior limbs of internal capsules
- Corpus callosum: Normal
- SPG30B: Paraplegia with Ataxia, mild & Sensory neuropathy, Recessive
56
● ATSV (KIF1A); Chromosome 2q37.3; Recessive or Dominant or de novo
- Epidemiology: Algerian & Pakistani families
- Genetics
- Inheritance: Recessive
- Mutations
- Missense: A255V, R350G
- Homozygous, often
- General location: Motor domain of kinesin 1A
- Allelic disorders
- ATSV protein
- Clinical: Pure or complicated SPG
- Onset
- Age: Mean 17.5 years; 12 to 21 years
- Spastic paraparesis
- Spastic gait
- Tendon reflexes: Increased in legs
- Plantar response: Extensor in 75%
- Sphincter involvement: Mild; 50%
- Functional: Inability to run
- Peripheral neuropathy
- Sensory loss: Legs; Panmodal; 50%
- NCV: Axon loss; Legs > Arms
- Ataxia: Mild
- Limb
- Eye: saccadic pursuit 50%
- Onset
- Laboratory
- CT scan: Cerebellar atrophy in 3 patient
- ATSV (KIF1A) variant: SPG30A Dominant or de novo
103
- Epidemiology: 27 families
- Genetics
- Inheritance: Dominant or Sporadic
- Mutations: S69L; T99M; G102S; R167C; I1127T
- Clinical: SPG, Pure or Complicated
- Onset age: 1 to 63 years; Intrafamilial variation
- Spasticity
- Paraparesis (Legs > Arms
- Tendon reflexes: Often brisk
- Plantar reflexes: Extensor
- Intellect: Normal
- Sensation: Normal
- Pes cavus
- Laboratory
- NCV: Axon loss or Normal
- Brain imaging: Uaually normal
- ATSV (KIF1A) variant: NESCAVS Dominant or de novo
- Nosology: NEurodegeneration, Spasticity ± Cerebellar Atrophy, Cortical Visual impairment Syndrome
- Epidemiology: > 40 patients
- Genetics
- Inheritance: Dominant or de novo
- Mutations: Missense
- ATSV (KIF1A) protein
- Clinical: Heterogeneous
- Onset: Infancy or Early childhood
- Developmental delay: Global, Walking, Speech, learning
- Spasticity: Legs; Loss of ambulation
- Visual impairment: Cortical; Optic atrophy, Oculomotor apraxia
- Polyneuropathy: Axonal, Dysautonomia
- Ataxia
- Extrapyramidal: Dystonia, Athetosis
- Skeletal: Scoliosis, Kyphosis, Joint contractures
- Course: Progressive
- Laboratory
- Brain MRI: Cerebellar atrophy, especially vermis; Corpus callosum thin
- SPG39: Spastic Paraplegia with Distal Arm & Leg Wasting
64
● Patatin-like phospholipase domain-containing protein 6 (PNPLA6; Neuropathy target esterase (NTE)); Chromosome 19p13.2; Recessive
- Epidemiology: Ashkenazi-Jewish & Northern European (German) families
- Genetics
- Mutations: Spastic ataxia
- Missense: Met1012Val & Arg890His
- Frameshift insertion: c.2946_2947insCAGC
- General location: N-terminal side of the EST domain (Gly840¢Ser1031)
- Allelic with
- Ataxia & Hypogonadism syndromes: Gordon¢Holmes; Boucher¢Neuhõuser
- Ataxia + Retinal changes
- Ataxia
- Oliver-McFarlane syndrome
- Laurence-Moon syndrome
- Mutations: Spastic ataxia
- PNPLA6 Protein
- Nosology: Also called Neuropathy target esterase & Neurotoxic esterase
- Family: Patatin-like phospholipase domain-containing proteins
- Involved in neuronal development
- Cell-signaling pathway controlling interactions between neurons and accessory glial cells
- Target for neurodegeneration induced by organophosphorus pesticides & chemical warfare agents
- Phospholipid esterase domain (EST): Altered by organophosphorous intoxication
- Can hydrolyze intrinsic membrane lipids
- Esterase activity: De-esterifies phosphatidylcholine
- Lysophospholipase activity: Catalysis of 2-arachidonoyl lysophosphatidylinositol
- Neurons: High expression
- Peripheral nerve
- Expression
- Mature non-myelinating Schwannn cells: Enriched areas
- Schmidt-Lanterman incisures
- Around Nucleus
- Myelin ovoids with axon degeneration
- Not in: Promyelinating Schwann cells
- Mature non-myelinating Schwannn cells: Enriched areas
- Required for: Ensheathment of unmyelinated axons (Remak fibers)
- Expression
- Mutations: Reduced NTE activity
- Clinical
- Onset
- Age: Childhood; ≤ 7 years
- Gait disorder: Spasticity
- Paraplegia
- Spastic
- Especially hamstrings
- Gait impairment
- Tendon reflexes
- Knees brisk
- Ankles often reduced
- Plantar response: Extensor
- Symmetric
- Progression: Slow
- Spastic
- Sensation reduced: Distal legs; Vibration
- Weakness: Hands; Iliopsoas; Tibialis anterior
- Muscle wasting: Distal hands & feet
- Urinary urgency
- Laboratory
- MRI: Spinal cord atrophy, especially thoracic
- NCV
- Motor axonopathy: Arms & Legs
- CMAP amplitudes: Reduced
- Sensory: Normal
- EMG: Denervation, Chronic & Ongoing
- PNPLA6 Variant syndromes: Ataxia & Hypogonadism 95
- Genetics
- Mutations: General location:
- C-terminal end of EST domain (Ser1031¢Pro1122)
- Inheritance: Recessive
- Allelic with: Spastic ataxia & SPG syndromes
- Mutations: General location:
- PNPLA6 protein
- Boucher-Neuhäuser syndrome
- Mutations: Missense or Stop
- Clinical
- Onset age: 1 to 6 years
- Visual impairmant
- Gait ataxia: Onset 6 to 27 years
- Hypogonadotropic hypogonadism
- Primary amenorrhoea
- Puberty: Delayed
- Chorioretinal dystrophy
- Spasticity: Few patients
- Muscle wasting, distal (50%)
- Tendon reflexes: Reduced
- Cognitive impairment: Mild
- Laboratory
- Brain imaging: Atrophy of cerebellum ± pons
- Gordon Holmes (Spastic) ataxia syndrome
: 1 patient
- Genetics
- Mutation: Frameshift; c.3084_3085insGCCA
- Other Gordon Holmes syndrome mutations: RNF216
- Clinical
- Onset
- Age: 14 years
- Delayed puberty
- Hypogonadotropic hypogonadism
- Spasticity: Legs
- Gait disorder: Onset age 30 years
- Onset
- Laboratory
- Brain imaging
- Cerebellar atrophy
- Empty sella
- Brain imaging
- Genetics
- PNPLA6 Variant syndrome: Cerebellar ataxia 108
- Epidemiology: 1 Zoroastrian (Parsi) family
- Genetics
- Inheritance: Recessive
- Mutation: D1310V; Homozygous
- Clinical
- Onset age: 2nd decade
- Ataxia
- Gait & Limb coordination
- Slow saccades
- Tendon reflexes: Brisk in legs
- Course: Slow progression
- Laboratory
- Brain MRI: Cerebellar & Midbrain artrophy
- NCV: Normal
Gordon Holmes Ataxia
Cerebellum: Control
Cerebellum: Patient
Medulla: PatientHolmes, Brain 1907;30:466-489
- Spastic paraparesis, Optic atrophy & Polyneuropathy (SPOAN)
52
● Kinesin light chain 2 (KLC2); Chromosome 11q13.2; Recessive
- Epidemiology: Brazilian families
- 70 patients
- Common in Serrinha dos Pintos (1/250; Carriers 1 in 9)
- 2 Egyptian siblings
- Genetics
- Mutation: chr11.hg19:g.66,024,557_66,024,773del; non-coding upstream region of the KLC2 gene
- Mutation effect: KLC2 over-expression
- Allelic, or same, disorder: SPG68
- Also see: SPOAN-like syndrome-IBA57
- KLC2 protein
- Kinesin light chain
- Kinesin protein-1 complex
- Binds to kinesins heavy chain in stoichiometric ratio of 1:1
- Expressed in Schwann cells & Axons
- Anterograde axoplasmic transport
- Kinesins
- Molecular motor
- Generates ATP-dependent movement of vesicles & organelles along microtubules
- Other disorders
- Clinical
- Onset: Infancy
- Spastic paraparesis
- Onset: Infancy
- Legs > Arms
- Motor delay: Toe walking; Some never walk
- Tendon reflexes: Increased proximally; Reduced distally
- Clonus: Spontaneous > Evoked
- Plantar responses: Often no response
- Progressive
- Optic atrophy
- Congenital
- Visual loss: Non-progressive
- Fixation nystagmus
- Pale optic disks
- Polyneuropathy
- Onset: Late childhood/early adolescence
- Motor & Sensory
- Distal amyotrophy: > 20 years of age
- Hyperhidrosis
- Vibration & joint position sense: Reduced
- Pain & temperature sensation: Normal
- Axonal
- Skeletal
- Joint contractures
- Spine deformity: Scoliosis
- Progressive
- Other
- ? Extrapyramidal: Speech disorders
- Dysarthria: Onset in 3rd decade
- Dysphonia
- Cogwheel rigidity (20%)
- Exacerbated acoustic startle response
- Cognitive: Normal
- Hearing: Normal
- ? Extrapyramidal: Speech disorders
- Course
- Wheelchair bound by 15 years
- Laboratory
- NCV: Axonal sensory-motor neuropathy
- Nerve biopsy
- Axon loss
- Occasional: Abnormally folded myelin; Onion bulbs
- Spine MRI: Mild cord atrophy
- Differential diagnosis: SPG 57
- Epidemiology: Brazilian families
- SPG68: See SPOAN
97
- Epidemiology: 2 patients, 1 family
- Genetics
- Original description: Mutations in FLRT1
- Re-assesment: KLC2 mutations
- Original description: Mutations in FLRT1
- Clinical: Complicated SPG
- Onset age: 2 to 3 years
- Gait: Disordered or Lost
- Tendon reflexes: Increased
- No spasticity
- Motor: Foot drop; Amyotrophy, mild
- Eye: Nystagmus, Optic atrophy
- Cognition: Normal
- Laboratory
- VEP: Abnormal
- MRI: Normal brain
- NCV/EMG: Polyneuropathy
- Hereditary sensory neuropathy with spastic paraparesis (Cavanagh's variant)
● Chaperonin containing T-complex polypeptide 1, subunit 5 (CCT5); Chromosome 5p15.2; Recessive
- Allelic with: HSN with Spastic paraparesis
- Clinical
- Sensory neuropathy
- Loss of pain & temperature
- Relatively preserved vibration & joint position sense
- Pathology: Axon loss, Small > Large fiber
- Acromutilation
- Spasticity
- Sensory neuropathy
- See: Hereditary sensory neuropathy
- HMSN 5
Spasticity + Ocular disorders
- Spastic paraplegia with retinal degeneration (SPG15)
:
- Spasticity with Pigmentary tapetoretinal degeneration & Mental retardation
: 15q24
- Sjögren-Larsson Syndrome
- Friedreich ataxia
- Spastic paraplegia, Optic atrophy, Microcephaly & XY sex reversal
- Infantile optic atrophy with chorea & spastic paraplegia (MGCA3)
: OPA3
- SPOAN
- Also see: Optic atrophy
Spasticity + CNS
- See: Spastic ataxia syndromes
- Friedreich ataxia
- Intermediate number of trinucleotide repeats
- Spastic ataxia
- Leukodystrophies:
MLD &
Krabbe
- Late onset disorders may present with spasticity & polyneuropathy
- DRPLA:
Homozygotes with intermediate # (40 or 41) of repeats
- Cerebrotendinous xanthomatosis
- Hyperornithinemia-hyperammonemia-homocitrullinuria (HHH syndrome)
- Cerebral palsy, Spastic Quadriplegic (CPSQ)
- Spastic paraplegia + Myoclonic epilepsy
● Autosomal Recessive- Onset: Prenatal to 10 years
- Spastic paraplegia
- Epileptic myoclonus
- Muscle atrophy: Distal
- Mental retardation
- Ataxia
- Hearing loss
- Course: Progressive
- SPG21: Spastic paraplegia with dementia & extrapyramidal signs (Mast syndrome)
42
● Acidic cluster protein, 33-KD (ACP33; Maspardin); Chromosome 15q22.31; Recessive
- Epidemiology: Ohio Amish; Japanese
- Genetics (ACP33 gene)
- Mutations
- Amish: Homozygous 601insA
- Japanese: Ala108Pro
- Mutations
- Maspardin protein
- Localizes to intracellular endosomal/trans-Golgi transportation vesicles
- May function in protein transport and sorting
- Clinical features
- Onset
- Age
- Amish: 2nd & 3rd decades
- Japanese: 6th & 7th decades
- Amish: Subtle changes in childhood
- Delayed motor milestones
- Awkward running
- Mild incoodination
- Age
- Spastic paraparesis
- Legs > Arms
- Gait disorder
- Jaw Jerk: May be brisk
- Bulbar
- Dysphagia: May need G-tube
- Speech disorders: Mild dysarthria
- Cortical function
- Personality/Psychiatric disorders: Shy; Nervous
- Dementia: May be severe
- End stage: Akinetic mutism
- Extrapyramidal signs: Late
- Rigidity
- Oromandibular dyskinesia
- Athetoid movements of the fingers or limbs
- Cerebellar ataxia: Late
- Tendon reflexes: Reduced at ankles
- Sensation: Normal
- Course: Slow progression
- Variant
- Japanese patients: No extrapyramidal, cerebellar, or bulbar signs
- Onset
- MRI
- Corpus callosum: Thin
- White-matter abnormalities
- Phenylketonuria: Adult onset Spastic paraparesis
+ Dementia
20
● Phenylalanine hydroxylase (PAH); Chromosome 12q23.2; Recessive
- Onset: 5th & 6th decade
- Spastic paraparesis
- Gait disorder
- Tendon reflexes: Brisk
- Dementia
- Strength: Normal
- Cranial nerves: Normal
- Fair Hair & Skin
- Treatment: Dietary restriction of phenylalanine
- SPG9A: Cataracts, Gastroesophageal Reflux, & Spastic Paraparesis with Amyotrophy
102
● delta-1-pyrroline-5-carboxylate synthase (ALDH18A1; P5CS); Chromosome 10q24.1; Dominant
- Epidemiology: Italian & British families
- Genetics
- Mutations: c.727G4C; c.755G4A
- Allelic disorders
- SPG Dominant: SPG9A
- SPG Complex or Pure (SPG9B): Dominant or Recessive
- Motor Neuronopathy with Cataracts & Skeletal abnormalities
- Cutis laxa, autosomal recessive, type IIIA
- Similar locus: Recessive SPG27
- Protein
- Clinical
- Onset
- Infancy to 4th decade
- Anticipation
- Cataracts (100%): Bilateral; Congenital
- GE Reflux (100%): Persistent vomiting; Hiatus hernia
- Spastic paraparesis
- Onset: 1st to 3rd decade; Some during pregnancy
- Penetrance: Incomplete
- Muscle wasting
- Hands & Forelegs
- Motor neuropathy with axon loss
- Skeletal: Growth impairment; Pes cavus
- Onset
- Laboratory
- MRI: Spinal cord atrophy
- Plasma Citrulline: Low
- SPG9B: Hereditary Spastic paraparesis, Complex or Pure
102
● delta-1-pyrroline-5-carboxylate synthase (ALDH18A1; P5CS); Chromosome 10q24.1; Recessive or Dominant
- Nosology: Aldehyde dehydrogenase 18 family, member A1
- Epidemiology: 7 families
- Genetics
- Mutations
- Dominant: Val120Ala; Arg252Gln; Ser652Phe; Arg665Leu
- Recessive: Asp715His; Arg128His, Leu637Pro
- Allelic disorders
- Mutations
- ALDH18A1 protein
- δ-1-pyrroline-5-carboxylate synthase (P5CS)
- Catalyses 1st common steps of arginine, proline & ornithine biosynthesis from glutamate
- Clinical
- Recessive
- Onset age: <1 to 7 years
- Cognitive impairment: Progressive
- Upper motor neuron
- Tetraparesis
- Pseudobulbar palsy
- Tendon reflexe: Increased in arms & legs
- Plantar response: Extensor
- Urinary incontinence
- Course: Progressive; Ambulation often lost
- Cerebellar: Postural tremor in arms (30%)
- Skeletal: Microcephaly; Facial dysmorphism; Short stature
- Dominant
- Onset age: Range 14 to 59 years
- Cramps
- Upper motor neuron
- Leg stiffness & Spasticity
- Spastic gait
- Tendon reflexes: Increased in arms & legs
- Plantar response: Extensor
- Dysarthria
- Weakness
- Distal > Proximal
- Motor neuropathy (45%)
- Sensory: Vibration loss in some
- Pes cavus (50%)
- Urinary symptoms (50%)
- Cognition: Normal
- Recessive
- Laboratory
- Amino acids: Blood
- Dominant families
- Citrulline: Low
- Proline, Ornithine, Arginine: Variably low
- Recessive: Normal
- Dominant families
- Amino acids: Blood
- SPG78: Spastic Ataxia
110
● ATP13A2;
Chromosome 1p36.13; Recessive
- Nosology: PARK-ATP13A2
- Epidemiology: Multiple families
- Genetics
- ATP13A2 protein
- Locations: Lysosomal; Late endosome; Endoplasmic reticulum
- P5-type subfamily of P-type transport ATPases
- Transporter
- Cations
- Polyamines: Especially spermidine
- Across membranes
- Endocytosis into cytosol
- Endolysosome system
- Interaction with heavy metals: In postmitotic neurons
- Expressed in brain
- ATP13A2 mutations: Polyamine-induced cell toxicity
- Lysosomal dysfunction & rupture
- Cathepsin B
activation
- Clinical
- Onset age: Juvenile & Adult
- Spastic quadraplegia
- Gait disorder
- Tendon reflexes: Increased
- Spasticity
- Plantar response: Extensor
- Loss of ambulation: 8 to 18 years after disease onset
- Cognitive
- Decline
- Psychiatric syndromes
- Pes cavus: Bilateral
- Eye
- Strabismus
- Supranuclear: Upgaze reduced; Gaze paresis
- Cerebellar
- Ataxia (30%): Limb; Gait
- Nystagmus, Lateral gaze
- Extrapyramidal
- Parkinsonism: May respond to levodopa
- Dystonia
- Polyneuropathy: Sensory-Motor
- Laboratory
- Brain MRI
- Cerebral & Cerebellar atrophy
- Basal ganglia disorder
- NCV: Axon loss
- Brain MRI
- ATP13A2 (PARK9) variant: ALS
122
- Epidemiology: 2 patients
- Genetics
- Mutations: p.Glu613Ter, p.Ile411Met
- Clinical
- Onset age: 4th to 5th decade
- Spasticity: Legs > Arms; Gait
- Tendon reflexes: Brisk
- Bulbar: 1 patient
- Tongue: Atrophy with Fasciculations
- Dysphagia
- Cognitive disorder: Mild in 1 patient
- Course: Progressive
- Laboratory
- Electrodiagnostic
- Motor unit potentials: Polyphasic
- Axonal sensory-motor polyneuropathy
- MRI: Normal
- Brain scans: Striatal pathololgy
- Electrodiagnostic
- SPG79B: Neurodegeneration with Optic atrophy, Childhood-onset (NDGOA); Spastic Ataxia
● Ubiquitin carboxyl-terminal esterase L1 (UCHL1; PGP9.5);
Chromosome 4p13; Recessive
- Epidemiology: Norwegian & Turkish families, 6 patients
- Genetics
- UCHL1 protein
- Hydrolyze: Small C-terminal adducts of ubiquitin to generate the ubiquitin monomer
- Expression: Neurons; Unmyelinated axons; Neuroendocrine cells, nomal & neoplasm; Testis/Ovary
- Binds to: Ubiquitin
- Mutation: Reduced enzyme activity
- Clinical
- Onset age: 5 to 10 years
- Vision loss: to blindness
- Upper motor neuron
- Spasticity
- Tendon reflexes: Increased
- Cerebellar ataxia
- Inability to stand
- Intention tremor
- Head tremor
- Nystagmus, gaze evoked
- Sensory: Vibration ± Position loss
- Fasciculations: 50%
- Strength: Normal or Reduced
- Cognition: Normal
- Pes cavus
- Course: Progressive
- Laboratory
- EMG: Myokymia
- NCV: Axon loss, Sensory & Motor
- Eye
- VEP: Reduced or Absent response
- Electroretinogram (ERG): Normal
- SSEP: Dorsal column dysfunction
- Brain MRI: Cerebral, Optic nerve & chiasm + Cerebellar atrophy
- Muscle biopsy: Normal or Chronic denervation
- Mouse model: gracile axonal dystrophy (gad)
- UCHL1 variant: SPG79A
- Epidemiology: 44 patients; 21 families
- Genetics
- Inheritance: Dominant
- Mutations: Loss of function
- Clinical
- Onset age: Median 49 years; Range Childhood to 70 years
- Ataxia: Gait disorder; Dysarthria
- Spasticity: Legs > Arms; Tendon reflexes brisk
- Some patients
- Optic atrophy
- Neuropathy: Sensory (Vibration loss); Axon loss
- Course: Slow progression
- Laboratory
- Brain MRI: Cerebellum small or normal
- Spastic paraplegia, Intellectual disability, Nystagmus & Obesity (SINO)
113
● Kinase D-interacting substrate, 220-kD; (KIDINS220/ARMS);
Chromosome 2p25.1; Sporadic, Dominant
- Nosology: ARMS (Ankyrin Repeat-rich Membrane Spanning)
- Epidemiology: 18 patients
- Genetics
- Mutations: de novo; Nonsense, Splice or Missense (Glu1223Gly); Heterozygous
- Mutation locations: Last two exons of gene
- Allelic Disorder: Ventriculomegaly & Arthrogryposis, Recessive
- KIDINS220 protein
- Scaffold protein
- Coordinates neurotrophin signal pathways in neurites: NGF; BDNF
- Controls axonal & dendritic maturation
- Neuronal survival, Axon guidance, Synaptic plasticity
- Mutation effect: Truncated isoforms
- Clinical
- Onset age: Congenital or Prenatal to 10 years
- Spastic paraplegia
- Spasticity: Legs
- Tendon reflexes: Increased
- Axial hypotonia (1 patient)
- CNS: Global delay, moderate
- Dysmorphism
- Brachyplagiocephaly
- Macrocephaly
- Forehead: Bossed or Prominent
- Eyes: Deep set
- Eyes
- Nystagmus
- Reduced acuity
- Squint
- Hypermetropia
- Astigmatism
- Obesity
- Peripheral neuropathy: 1 patient
155
- Motor > Sensory
- Axon loss
- Course
- Long survival
- Slow progression
- Laboratory
- Brain imaging
- Lateral & 3rd ventricle dilation
- White matted reduced
- Atrophy, mild
- Nerve: Axon loss ± swelling
- Brain imaging
- KIDINS220 variant: Ventriculomegaly & Arthrogryposis, Recessive (VENARG)
- Epidemiology: 3 families
- Genetics
- Inheritance: Recessive
- Mutations: Frame-shift or In-frame deletion
- Clinical
- Prenatal
- Ventriculomegaly
- Arthrogryposis
- Face: Dysmorphic
- Subcutaneous edema
- Cardiac defects: Some patients
- Death: Intrauterine or Childhood
- Pathology
- Cortical & Cerebellar hypoplasia
- Corpus callosum: Thin
- Spastic Paraplegia, Complicated (SPG81)
114
● Ethanolaminephosphotransferase 1 (EPT1; Selenoprotein I; SELENOI)
;
Chromosome 2p23.3; Recessive
- Epidemiology: 2 Arab family, 5 patients
- Genetics
- Mutations: c.335G>C (p.Arg112Pro); c.732-2A-G (Splice site)
- EPT1 protein
- Final step in phosphatidylethanolamine synthesis
- Kennedy pathway phospholipid cascade
- Clinical
- Onset age: Infancy/Early childhood
- Motor
- Delayed gross motor development<
- Motor function: Gradual decline
- Dysarthria
- Spastic paraperesis
- Spasticity
- Tendon reflexes: Increased
- Plantar response: Extensor
- Progressive
- Arms involved later in course
- Peripheral neuropathy (Oldest patient): Axon loss; ?? Demyelinating
- Cortical & CNS
- Intellectual impairment
- Language delay
- Microcephaly
- Seizures
- Eye
- Retinal pigment disorder
- Cone-rod dysfunction
- Visual acuity: Reduced
- Bifid uvula
- Laboratory
- Brain MRI: Increased T2 signal intensity in periventricular white matter
- NCV: CMAPs normal or reduced amplitude & dispersed
- Cataracts, Spastic Paraparesis & Speech Delay (CSPSD)
● Fatty Acyl CoA Reductase 1 (FAR1);
Chromosome 11p15.3; Dominant
- Epidemiology: 12 patients
- Genetics
- Mutations: Arg480 residue; Heterozygous
- Allelic disorder: Peroxisomal fatty acyl-CoA reductase 1 disorder
- FAR1 protein
- Converts fatty acyl-CoA into a fatty alcohol & CoASH
- Wax monoester synthesis
- Mutations: Loss of plasmalogen-dependent feedback on FAR1 protein levels
- Clinical
- Onset age: Early childhood
- Spastic paraparesis
- Seizures (65%)
- Speech delay (80%)
- Cataracts: Bilateral; Often congenital
- Constipation (45%)
- Course: Ambulatory, but requiring walking aids
- Laboratory
- Brain imaging: Usually normal
- SPG, Complex with Ataxia
141
● RAD50-interacting protein 1 (RINT1);
Chromosome 7q22.3; Recessive
- Epidemiology: 3 patients
- Genetics
- Mutations: Stop or Deletion
- Allelic disorder: Infantile liver failure syndrome 3
- RINT1 protein
- Autophagy
- Lipid homeostasis: Neutral & Phospholipid metabolism
- Mitochondrial dynamics
- Regulation of membrane traffic between the Golgi & Endoplasmic reticulum (ER)
- Telomere length control
- Clinical: Spastic-Ataxia
- Onset age: Early childhood
- Motor & Intellectual development: Delayed
- Spasticity
- Ataxia: Gait; Dysmetria; Nystagmus
- Optic atrophy
- Strabismus
- Dysmorphic face: Low set ears
- Hepatitis
- Laboratory
- Brain MRI
- Posterior periventricular white matter hyperintensity
- Corpus callosum: Thin
- Cerebellar atrophy
- Optic chiasm atrophy
- Hepatic dysfunction
- Brain MRI
- Spastic paraplegia, Epilepsy & Mental retardation
36
● Autosomal Dominant- Onset: < 40 years
- Spastic paraplegia: Legs > Arms
- Epilepsy: At presentation
- Mental retardation: Variable
- Lawrence-Moon syndrome
● PNPLA6; Chromosome 19p13.2; Recessive- Genetics
- Clinical
- Spastic parapaesis
- Pigmentary retinopathy
- Mental retardation
Cerebral palsy, Spastic Quadriplegic (CPSQ)
|
CPSQ1 (DEE89): GAD1; 2q31 CPSQ2: KANK1; 9p24 CPSQ3: ADD3; 10q25 CPSQ4 (SPG51): AP4E1; 15q21 CPSQ5 (SPG47): AP4B1; 1p13 CPSQ6 (SPG52): AP4S1; 14q12 CPSQ (SPG50): AP4M1; 7q22 NEDSBAS: WDR45B; 17q25 NEDSGA: GRIA4; 11q22 NMIHBA: PRUNE1; 1q21 SLS-like: ELOVL4; 6q14 SPATCCM: SLC1A4; 2p14 Cerebral palsy, Ataxic KCNC3 ITPR1 SPTBN2 |
CPSQ1: Developmental and epileptic encephalopathy 89 (DEE89)
● Glutamate decarboxylase 1 (GAD1; GAD67)
- Genetics
- Mutation: Ser12Cys
- Allelic disorder: Developmental and epileptic encephalopathy 89
- Epidemiology
- Pakistani families
- No recognizable adverse pre- or postpartum events
- GAD1 protein
- Catalyzes conversion of glutamic acid to gamma-aminobutyric acid (GABA)
- Clinical
- Onset age: Infancy
- Symmetry of neurological signs
- Spasticity: Legs > Arms; Tendon reflexes increased
- Cortical: Mental retardation; Seizures, Occasional
- Microcephaly: Some patients
- Scoliosis
CPSQ2
● KN motif and ankyrin repeat domains 1 (ANKRD15; KANK1)
- Epidemiology: Israeli family of Jewish Moroccan origin
- Genetics
- Mutation: Deletion containing only ANKRD15 gene
- Inheritance dependent on parental gender: All affected children born to related clinically healthy fathers
- Paternal transmission (to affected children): Region is hypomethylated in child
- Maternal transmission: Region is hypermethylated
- Affected patients: Have deletion from father & maternal allele with gene repressed from mother
- Allelic disorders: KANK1 mutations increased (but low) frequency in ALS vs controls 132
- ANKRD15
- Ubiquitous expression
- Component of AP-4 complex
- Involved in the recognition and sorting of cargo proteins with tyrosine-based motifs
- From trans-golgi network to endosomal-lysosomal system
- KANK1: Related to ALS cytoskeletal genes (PFN1, KIF5A, TUBA4A)
- Clinical
- Onset
- Congenital
- Hypotonia
- Spastic quadriplegia: Evolves over 1st year
- Mental retardation
- Nystagmus: Transient
- Onset
- Laboratory
- MRI: Brain atrophy; Ventriculomegaly
Spastic Paraplegia 50, Recessive (CPSQ3; SPG50)
● Adaptor-related protein complex 4, Mu-1 subunit (AP4M1; MU-ARP2)
- Epidemiology: 60 families
- Genetics
- Mutation: Stop
- AP4 proteins
- Clinical
- Onset
- Infancy: Ventricular diatation in 20th week of pregnancy
- Hypotonia
- Strabismus
- Spastic quadriplegia
- Progressive over 1st year
- Pseudobulbar signs
- No ambulation
- Hypertonia, generalized
- Hyperreflexia
- Plantar responses: Extensor
- Sphincter control: Absent
- Mental retardation: Absent speech
- Microcephaly
- Adducted thumbs
- Course: Minimal change after 1st year
- Onset
- Laboratory
- MRI: Ventriculomegaly; White matter changes; Cerebellar atrophy
- Pathology
- Reduced myelin
- Gliosis
- Dendritic arborization
- Abnormal morphology
- Aberrant GluRdelta2 glutamate receptor localization
Cerebral Palsy with Microcephaly & Intellectual Disability 4 (CPSQ4; SPG51)
● Adaptor-related protein complex 4, Epsilon-1 subunit (AP4E1)
- Epidemiology: 25 families
- Genetics
- Mutations: Nonsense, Deletion, Insertion; Stop
- Allelic disorder: Stuttering, familial persistent, 1 (Dominant) (STUT1)
- AP4 proteins
- Clinical
- Spastic cerebral palsy
- Early infancy: Hypotonia
- Tetraplegic
- Tone: Increased
- Tendon reflexes: Increased
- Plantar reflex: Extensor
- Intellectual
- Disability: Severe
- Speech: Very dleayed
- Laughter: Stereotypic
- Microcephaly
- Epilepsy
- Eye & Ear: Normal
- Course: Live to adulthood
- Spastic cerebral palsy
- Laboratory
- MRI: Dilated ventricles; Cerebellar atrophy; White matter abnormal
Cerebral Palsy with Microcephaly & Intellectual Disability 5 (CPSQ5; SPG47)
● Adaptor-related protein complex 4, Beta-1 subunit (AP4B1)
- Epidemiology: 53 families
- Genetics
- Mutations: Truncating
- Allelic with: SPG 47
- AP4B1 protein
- Subunit of heterotetrameric adaptor protein (AP) complex
- Intracellular transport of proteins: Especially neurons
- Adaptor protein complexes (AP1-4)
- Expression: Ubiquitous
- Heterotetrameric complexes
- Mediate
- Vesicle formation
- Selection of cargo molecules for inclusion into vesicles
- Brain development
- AP4 disorders
- Clinical
- Onset age: Birth
- Microcephaly
- Muscle tone: Hypotonic early; Hypertonic with development
- Spastic paraplegia
- Inability to walk unaided
- Hyperreflexia
- Waddling gait
- Other CNS
- Cognitive deficit: Severe
- Speech delay
- Adaptive impairment
- Stereotypic laughter
- Shy character
- Skeletal
- Palate: High
- Nasal bridge: High
- Short stature
- Joints: Hyperlaxity
- Genu recurvatum
- Pes planus
Spastic paraparesis with Intellectual Disability, Shy Character & Short Stature (CPSQ6; SPG52)
● Adaptor-related protein complex 4, Sigma-1 subunit (AP4S1)
- Epidemiology: 26 families
- Genetics
- Mutations: Nonsense; Frameshift; Splice
- AP4 proteins
- Clinical
- CNS
- Intellectual disability, severe
- Speech: Absent
- Shy character
- Stereotypic laughter
- Motor
- Muscle mass in legs: Reduced
- Inability to walk
- Early: Hypotonia
- Late: Spastic paraparesis
- Skeletal
- Microcephaly
- Foot deformity
- Growth retardation
- CNS
Cerebral palsy, Spastic quadriplegic (CPSQ3)
● Adducin 3 (ADD3; ADDL; Gamma adducin)
- Epidemiology: 1 Jordanian family, 4 patients
- Genetics
- Mutation: Homozygous; Missense; G367D
- Adducin 3 protein
- Expression: Ubiquitous
- Actin-capping
- Modulate fast-growing end of actin filaments
- Control actin molecule length
- Upregulated in chemoresistant glioblastoma cells
- Adducin family
- α-Adducin (ADD1)
: Roles in maintaining axon diameter & endocytosis
- α-Adducin (ADD1)
- Clinical
- Onset age: Early childhood
- Spastic quadriplegia: Moderate to Severe
- Pyramidal signs
- Dysarthria
- Dysphagia
- Cognitive impairment
- Eye: Exotropia, Supranuclear gaze palsy, Strabismus, Nystagmus
- Seizures: 1 patient
- Microcephaly: Borderline
- Laboratory
- Brain MRI
- T2-weighted hyperintensities: Grey matter heterotopia
- White matter reduced, especially frontoparietal
- May be normal
- Muscle biopsy
- Type 2 muscle fiber atrophy
- Mitochondrial oxidative enzymes: Complex I activity mildly reduced
- Brain MRI
Neurodevelopmental Disorder with Spastic Paraplegia & Microcephaly (NEDSPM)
● Glutamate Pyruvate Transaminase 2 (GPT2)
- Epidemiology: 8 families
- Genetics
- Mutations: Missense & Stop
- GPT2 protein
- Alanine transaminases
- Pyridoxal enzyme
- Catalyzes: Reversible transamination between alanine & 2-oxoglutarate to form pyruvate & glutamate
- High expression: Muscle, Fat, Kidney, Brain
- Clinical
- Onset: Infancy & Early childhood
- Hypotonia
- Developmental delay: Global
- Spasticity: Legs > Arms; Gait disorder
- Seizures
- Bulbar: Dysarthria; Drooling
- Microcephaly
- Course: Progressive
- Laboratory
- Brain imaging: Normal or Corpus callosum thin
- Alanine: Reduced in CSF
Neurodevelopmental Disorder with Spasticity & Thin corpus Callosum & Decreased White Matter (NEDSCW)
● Ribosomal Protein S6 Kinase C1 (RPS6KC1; RPK118)
- Epidemiology: 13 patients, 8 families
- Genetics
- Mutations: Missense & Stop
- RPS6KC1 protein
- Sphingosine kinase-1 (SPHK1)
-binding protein
- Sphingosine-1-phosphate (SPP)-mediated signaling
- Sphingosine kinase-1 (SPHK1)
- Clinical
- Head: Microcephaly
- Face: Dysmorphic; Long, Micrognathia
- Eyes: Hypotelorism; Strabismus; ± Ocular apraxia
- Spine: Scoliosis
- CNS
- Developmental delay: Motor & Intellect
- Spastic paraplegia
- Ataxia: Gait; Tremor
- Behavior: Autistic; Agression; Anxiety
- PNS: Axon loss in 1 patient
- Laboratory
- Brain imaging
- Corpus callosum: Thin
- White matter volume: Loss
- Cerebellar vermis: Atrophy
- Brain imaging
Neurodevelopmental disorder + Motor regression, progressive Spastic Paraplegia & Oromotor dysfunction (NEDRSP)
● Small nuclear RNA-activating protein complex, polypeptide 4 (SNAPC4)
- Epidemiology: 8 families
- Genetics
- Mutations: Loss of function
- SNAPC4 protein
- Subunit of snRNA-activating protein complex (SNAPc)
- Transcription of RNA polymerase II & III snRNA genes
- Clinical
- Onset ages: 6 to 36 months
- Spasticity: Appendicular
- Cognitive: Impaired
- Ataxic gait: Some patients
- Oromotor dysfunction: Slow speech, Dysarthria, Drooling
- Ocular: Nystagmus, Optic nerve pallor, Strabismus, Astigmatism, Oculomotor apraxia, Retinopathy
- Skeletal: Short stature; Microcephaly
- Laboratory
- Brain imaging; Cerebellar & Cerebral atrophy
Neurodegeneration, early-childhood-onset + Retinitis pigmentosa, Sensorineural hearing loss, & Demyelinating peripheral neuropathy (CONDRHN)
● Kinesin light chain 4 (KLC4)
- Epidemiology: Turkish family; 3 siblings
- Genetics
- Mutation: 19-bp deletion; Homozygous
- KLC4 protein
- Expressed in CNS & PNS
- Cytoplasm & Mitochondria
- Stabilization of nascent axon branches
- Microtubule dynamics
- Endosomal transport
- Clinical
- Onset age: 3 years
- Gait disorder, progressive walking difficulties
- Eye: Vision loss; Nystagmus; Optic palor
- Sensorineural hearing loss
- Intellectual development: Impaired
- Spasticity
- Weakness: Legs
- Laboratory
- NCV: Demyelinating neuropathy
- Serum lactate: High
- Brain MRI
- Abnormal dentate, posterior internal capsule, Subcortical white matter
- Cerebral & cerebellar atrophy
- Corpus callosum: Thin
Spastic tetraplegia, Thin corpus callosum & Progressive microcephaly (SPATCCM)
● Solute carrier family 1 (Glutamate/Neutral amino acid transporter), Member 4 (SLC1A4)
- Epidemiology: Ashkenazi Jewish & Pakistani families
- Genetics
- Mutations: c.944_945delTT; E256K (Ashkenazi); Arg457Trp (Pakistani)
- SLC1A4 protein
- Neutral amino acid transporter (ASCT1)
- L-serine, L-alanine, L-cysteine, L-threonine
- Brain: L-serine
- Synthesized by astrocytes
- Shuttled into neurons by SLC1A4 transporter
- Neutral amino acid transporter (ASCT1)
- Clinical
- Onset: Birth
- Psychomotor development delay: No speech; Inability to walk,
- Microcephaly: Postnatal, Progressive
- Spasticity: Legs > Arms
- Febrile seizures: Infancy; 1 patient
- Laboratory
- Brain MRI
- Corpus callosum: Thin
- Myelination: Delayed
- White matter: Nonspecific abnormalities
- Brain MRI
Neurodevelopmental disorder ± Seizures & Gait abnormalities (NEDSGA)
● Glutamate receptor, ionotropic, AMPA 4 (GRIA4)
- Epidemiology: 5 patients
- Genetics
- Mutations: de novo; Heterozygous; Missense
- GRIA4 protein
- L-glutamate-gated ion channel
- Mediate fast synaptic excitatory neurotransmission
- Responsive to glutamate agonist alpha-amino-3-hydroxy-5-methyl-4-isoxazolpropionate (AMPA)
- Clinical
- Onset
- Age: Neonatal
- Irritability
- Stiffness & Hypertonia
- Startle reflex: Increased
- CNS
- Seizures
- Spastic quadriplegia: Gait disorder
- Choreiform movements
- Developmental delay: Absent speech, Inability to follow commands
- Other: Some patients
- Sleeping problems
- Feeding difficulties
- Optic nerve hypoplasia
- Stereotypic hand movements
- Eye: Nystagmus, Strabismus
- Skeletal: Contractures, Short stature, Microcephaly
- Onset
- Laboratory
- Brain MRI: Cortical atrophy; Corpus callosum thin
Neurodevelopmental Disorder with Spastic Quadriplegia & Brain abnormalities ± Seizures (NEDSBAS)
● WD Repeat-containing protein 45B; (WDR45B)
- Epidemiology: 3 families, 6 patients
- Genetics: Stop mutations
- WDR45B protein
- WD40 repeat protein
- Regulate assembly of multiprotein complexes
- Presenting β-propeller platform for simultaneous & reversible protein-protein interactions
- Clinical
- Onset: Neonatal
- Cortical: Developmental delay; Intellectual disability
- Seizures: Some patients
- Spastic quadriplegia
- Axial hypotonia
- Skeletal: Contractures, Kyphoscoliosis, Microcephaly
- Course: Progressive
- Laboratory
- Brain MRI: Large ventricles, Thin cortex & Corpus callosum, Hypoplastic cerebrum, Reduced white matter
Neurodevelopmental Disorder with Spastic Quadriplegia, Hypotonia & Seizures (NMIHBA)
● Prune Exopolyphosphatase 1 (PRUNE1)
- Epidemiology
- 64 patients
- Neurodegeneration, childhood, in Manitoba Cree families
- Genetics
- Mutations
- Missense, Stop, Splice
- Most: Loss of function
- Manitoba Cree: c.521-2A>G
- Genotype-phenotype correlation: Poor
- Allelic disorder: Metastatic subgroups of medulloblastoma
- Mutations
- PRUNE1 protein
- Aspartic acid-histidine-histidine (DHH) protein superfamily
- Phosphoesterase
- Functions
- Nucleotide phosphodiesterase
- Exopolyphosphatase
- Induction & Regulation of cell motility & migration
- Role in brain development
- Cancer cell metastasis
- Mutations: Abnormal cytoskeletal rearrangements & Microtubule polymerization
- Clinical
- Onset age: Congenital to 3 months
- Spasticity: Quadriparesis; No independent ambulation
- Developmental delay: Global, No speech
- Seizures (80%)
- Dysphagia
- Skeletal: Scoliosis; Joint contractures
- Microcephaly: Some patients
- Polyneuropathy (50%): Demyelinating
- Tendon reflexes: Reduced or Absent in some patients
- NCV: Slow in some patients (15 to 29 M/s)
- Course: Progressive; Death in many < 20 years
- Laboratory
- Brain MRI: Cerebral atrophy; Brainstem lesions, T2 hyperintense
- Muscle: Denervation/Reinnervation
- Brainstem: β-Amyloid precursor protein inclusions
- Spinal cord: Motor neuron loss
Familial Spastic Paraplegia: X-linked
|
Adrenomyeloneuropathy: ALDP; Xq28 Mental retardation Infantile spasms: ARX; Xp22 Psychosis, & Macroorchidism: Xq28 Rett syndrome: MECP2; Xq28 Small testes: ATRX; Xq13 SPG1: L1CAM; Xq28 SPG2: PLP; Xq21 Strabismus: X Woods-Black-Norbury Syndrome: Xq26 SPG 16: Xq11 SPG 22: SLC16A2; Xq13 SPG 34: Xq25 |
- SPG1
● L1 Cell Adhesion Molecule (L1CAM)
;
Chromosome Xq28; Recessive
- L1 syndrome epidemiology: 1/3000¢1/25,000 live birth males
- Gene features
- Mutations
- > 300 described
- Multiple types: Missense, Nonsense, Splice, Small deletions
- Locations: All along gene; No hotspots
- Gene-Clinical correlations
- Truncation: Earlier death than missense
- Allelic disorders
- SPG1
- MASA Syndrome
- Mental Retardation; Adducted Thumbs; Shuffling Gait; Aphasia
- CRASH syndrome
- Corpus callosum hypoplasia, mental Retardation, Adducted thumbs, Spastic paraplegia & Hydrocephalus
- X-linked hydrocephalus
- X-linked partial corpus callosum agenesis: Missense mutations in extracellular domain
- Gene mutation data base: HGMD
- Mutations
- L1CAM protein
- Transmembrane glycoprotein
- Immunoglobulin (Ig) superfamily
- Development
- Neurite outgrowth guidance
- Neuron cell migration
- Axon bundling
- Synaptogenesis
- Myelination
- Long-term potentiation
- Malignancy: Progression & Metastatic cascade
- Clinical
- Spasticity: Paraplegia
- Ataxia
- Mental Retardation: Often severe
- Muscle: Absent extensor pollicis longus
- Female carriers: May have adducted thumbs & learning disability
- Laboratory
- Brain imaging: Corpus callosum abnormal; Hydrocephalus
- SPG2
137
● Proteolipid protein 1 (PLP1; Lipophilin)
;
Chromosome Xq22.2; Recessive
- SPG2 Epidemiology: 39 families
- Genetics
- Gene: Highly conserved among species
- Mutations
- PLP1: > 390 described
- SPG2: 35 mutations
- Spastic paraparesis: Point mutations (His139Tyr, Ser169Phe, Ile186Thr, Phe236Ser)
- Gene duplication
- Most common mutation (50% to 75%) in Pelizaeus Merzbacher
- Milder phenotype: Slow progression; No dementia
- Originates more 9 times frequently in male germ cells
- Duplicated gene may be inserted in variant locations on X-chromosome
- Point mutation: 15% to 20% of mutations
- Gene deletion
- Mechanisms
- Insertional translocation event
- Sister-chromatid exchange: In male meiosis
- Complex genomic rearrangement
- Clinical: PMP null syndrome
- Mechanisms
- Genetic duplication downstream of PLP gene: May silence PLP gene
- Allelic variants
- Pelizaeus-Merzbacher
- Hypomyelination of Early Myelinating Structures (HEMS)
- Proteolipid protein null syndrome
- PLP gene duplication syndrome ± Neuropathy
- SPG2 + Axonal neuropathy
- Multiple sclerosis: Predisposition
- Autism
- Neurodevelopmental disorders
- Early onset neurological disease trait (EONDT)
- Female SPG2: PLP1 mutation heterozygotes
- 16 patients described
- Gait disorders: Late onset
- EONDT: Large deletions
- Animal disorders
- Murine: Jimpy (Ala38Ser)
- PLP lacks amino acids 208-232; Hypomyelination; Absent mature oligodendrocytes
- Murine: Rumpshaker (Ile186Thr); Hypomyelination; Normal oligodendrocytes & longevity
- Rat: Myelin deficient (MD; Thr75Pro); Severe phenotype; PLP & DM20 both abnormal
- Chinchilla rabbits: Paralytic tremor (pt); CNS hypomyelination
- Murine: Jimpy (Ala38Ser)
- PLP1 protein
- Properties: Very hydrophobic; Integral membrane
- Most abundant protein in CNS myelin (50%)
- Roles
- Formation & maintenance of multilamellar structure of myelin
- Stabilize & Maintain myelin sheath and in the development
- Oligodendrocyte precursor development
- DM20 from alternative splicing of PLP gene: Affected in some families
- PLP1 deletions
- Physically fragile myelin sheath
- Susceptibility to subsequent demyelination
- PLP1 overexpression
- Myelination: Abnormal
- Oligodendrocytes: Reduced viability
- Clinical
- General
- Complicated SPG: 80%
- Onset
- Usually 1 to 5 years
- Varies with
- Allelic variant
- Specific mutation
- Sex
- Males: 2 years to Adult
- Females: Later than males in same family
- Gait disorder
- Spasticity
- Paraparesis: Legs > Arms
- Gait disorder
- Bladder
- Tendon reflexes: Increased
- Plantar reflex: Extensor
- Cognition: Normal or Cognitive disorders
- Progression
- Slow
- Life span: Normal
- Additional CNS signs: Present after paraparesis; Varies according to mutation
- Cognitive: Mental retardation (Common); Dementia (Occasional)
- Optic atrophy
- Cerebellar
- Ataxia
- Tremor: Kinetic; Arms > Legs
- Nystagmus
- General
- Laboratory
- Demyelinating neuropathy
- Clinical: Only mild manifestations
- NCV: Demyelinating
- Abnormal in 64%
- Motor nerve conduction velocity Δ
- Slow
- Non-uniform
- Motor > Sensory Δ
- Distal motor & F wave latencies: Prolonged
- MRI
- CNS myelination: Deficient; Leukoencephalopathy
- WM lesions: Periventricular, Parieto-occipital, Internal capsule, Corpus callosum, Subcortical, Medulla, Brainstem
- Pathology: Dysmyelination
- Demyelinating neuropathy
- Allelic variant syndromes
- Connatal Pelizaeus-Merzbacher
- Genetics
- Mutations produce misfolding of PLP & DM20
- Proteins accumulate in ER
- Clinical: Severe CNS changes
- Onset: Birth
- Hypotonia
- Spasticity: Severe
- Cerebellar: Ataxia; nystagmus
- Cognitive disorders
- No peripheral neuropathy
- Genetics
- Proteolipid protein null syndrome
142
- Epidemiology: > 20 patients
- Genetics: Mutation types
- Total PMP gene deletion
- Mutation in exon 1 (start codon)
- Deletions: 4th G of PLP coding region; Distal enhancer region
- PLP1 Protein Δ: Absent PLP/DM20
- Clinical
- Onset age: 1 to 5 years
- Motor development: Delay
- Intellectual disability
- Spastic paraplegia (70%)
- Ataxia
- Peripheral neuropathy
- Clinically mild
- Demyelinating
- Mild cognitive change
- Gastroesophageal reflux
- Survival: 5th to 7th decade
- Laboratory
- Brain MRI: Hypomyelination
- NCV: Demyelinating neuropathy (70%)
- Typical Pelizaeus-Merzbacher (HLD1)
- Genetics: Most often gene duplication
- External link: Gene clinics
- PLP gene duplication syndrome + Neuropathy
15
- Protein: Increased PLP/DM20
- Clinical
- CNS: Moderate to severe disease
- Peripheral neuropathy: Some patients
- Weakness: Hands & Feet
- Sensory loss ?
- Tendon reflexes: Reduced at ankles
- NCV: Absent sensory potentials
- Nerve pathology: Loss of large myelinated axons
- SPG2 + Axonal neuropathy: Genetic duplication downstream from PLP gene
55
- Clinical
- Gait: Awkward, Frequent falls, Steppage
- Dysarthria
- Muscle atrophy
- Weakness: diffuse; distal > Proximal
- Spastic paraplegia
- DTRs: Absent at ankle jerks
- Dystonic posture: With arms outstretched
- Mental retardation: Mild, Since infancy
- NCV
- Motor & Sensory amplitudes: Reduced
- Conduction velocity: Normal
- Nerve biopsy
- Reduced numbers of large & small myelinated axons
- Spheroids: Accumulations of normal and large sized mitochondria & tubulovesicular profiles
- Clinical
- Adrenomyeloneuropathy (AMN)
● Adrenoleukodystrophy protein (ALDP; ABCD1)
;
Chromosome Xq28; Recessive
- Epidemiology
- Incidence: 1: 20,000 to 1:100,000
- Genetics
- Multiple point mutations & deletions (6%): > 100 different
- No correlation with phenotype
- Allelic with
- Adrenoleukodystrophy
- Other syndromes
- ? Modifier gene: Some differences in phenotypes in identical twins
- ALDP protein
- ATP binding transporter: ABC transporter superfamily
- Location: Peroxisomal membranes
- Absent in 70% of X-ALD patients: No correlation with phenotype
- AMN: Clinical features
- Onset
- Age: 2nd to 6th decade
- Penetrance: 75% by 60 years
- Spastic paraparesis: Mainly legs
- Motor: Spastic gait; Increased tone in legs
- Sensory loss: Panmodal with proprioceptive loss
- Tendon reflexes: Increased in legs
- Neuropathy
- Specific clinical signs due to neuropathy uncertain
- Usually primary axonal degeneration
- Reduced large & small myelinated axons
- Little sprouting
- Unmyelinated axon involvement: Common
- Demyelinating changes: Mild
- Occasional onion bulbs reported
- Thinly myelinated axons: Some
- Schwann cell inclusions: Pi granules
- Autonomic
- Impotence: May be preservation of ejaculation & Orgasm
- Bladder: Urgency; Frequency; Incontinence
- Adrenal insufficiency:
1° adrenocortical insufficiency (70%)
- Weakness
- Weight loss
- Skin: Hyperpigmentation
- Nausea & Vomiting
- Gynecomastia
- Testicular atrophy
- Prognosis
- Progression to wheelchair over 5 to 15 years
- Better with normal brain MRI (50%): Less cognitive impairment
- Worse with cerebral involvement
- All X-ALD patients who survive to adulthood eventually develop AMN
- Treatment
- Adrenal insufficiency: Steroid replacement
- Neurological: None; Dietary therapy or marrow transplant not clearly effective
- Female carriers
- Myelopathy
- Age: Later onset; 20 to 55 years
- Frequency: 20% to 50%
- Myelopathy
- Onset
- Laboratory
- Electrophysiology
- Primary axonal neuropathy
- Variable demyelinating features: Male > Female
- Somatosensory evoked potentials
- Males: Central & Peripheral pathway changes
- Females: Central changes
- BAERs: Abnormal in males (100%) & females (50%)
- CSF: Normal until cerebral involvement
- Biochemistry
- Increased saturated, unbranched very long-chain (> 24 carbon) fatty acids (VLCFA)
- VLCFA accumulation
- Location: In all tissues
- No change with age
- 2° to: Defective formation of Coenzyme A derivative during oxidation
- C26:0 VLCFA may impair mitochondrial Complex V function
- Other peroxisomal syndromes with increased VLCFA: Zellweger; Infantile Refsum
- Carrier females: 85% with increased VLCFA
- Pathology: 2 types
- Distal axonopathy: 'Pure' AMN
- Inflammatory demyelinating CNS: Rapidly progressive cerebral forms
- Electrophysiology
- Other ALDP syndromes
- Childhood cerebral adrenoleukodystrophy
- Onset < 10 years
- Cognitive deficits
- CNS demyelination: Inflammatory
- Disability: Often < 3 years
- Adolescent cerebral: Onset 10 to 21 years
- Adult cerebral: Rapid CNS progression; Onset > 21 years
- Addison's disease
- Heterozygotes
- Mineralocorticoid insufficiency: Isolated
- Nonsteroidal anti-inflammatory agents: Cause hypoaldosteronism
- Glucocorticoid reserve: Subclinical reduction
- Adrenal cortical insufficiency: Rare
- Asymptomatic
- Childhood cerebral adrenoleukodystrophy
- Epidemiology
- Spastic diplegia with Mental retardation & Small testes (MRXS3; Sutherland-Haan)
● ATRX;
Chromosome Xq21.1; Recessive
- ATRX (Helicase 2)
- Location: Nuclear
- Function: Regulation of gene expression
- ATRX mutations also produce
- α-Thalassemia/Mental retardation syndrome
- Juberg-Marsidi mental retardation
- α-Thalassemia/Mental retardation syndrome
- Clinical
- CNS: Mental retardation
- Skeletal: Short stature; Microcephaly; Brachycephaly
- Testes: Small
- Spastic diplegia
- ATRX (Helicase 2)
- SPG 16: Spastic paraplegia, Pure
12
● Chromosome Xq11.2; Recessive- Genetics: Nucleus organizer region (NOR) insertion at Xq11.2
- Clinical
- Walking: Late or None
- Spastic paraplegia
- Spasticity: Legs
- Tendon reflexes: Brisk in lower extremities
- SPG 22: Allan-Herndon-Dudley Syndrome (AHDS)
● Solute Carrier Family 16 (Monocarboxylic acid transporter), Member 2 (SLC16A2; MCT8);
Chromosome Xq13.2; Semi-Dominant
- Genetics
- Mutations: Missense & Deletions (In & Out of frame)
- SLC16A2 protein
- Location: Choroid plexus & ventricular epithelium
- Iodothyronine transport
- Clinical
- Onset age: Birth
- Head & Neck
- Microcephaly
- Face: Elongated; Bitemporal narrowing
- Ears Large; Simple; Pinna abnormal; Antihelix prominent & flat
- Eyes: Disconjugate movements; Nystagmus, rotary (Some patients)
- Chest: Pectus excavatum
- Skeletal
- Contractures: Large & Small joints
- Scoliosis
- Feet: Flat; Great toe lateral deviation
- Neurologic
- Neonatal: Hypotonia
- Mental retardation
- Spasticity
- Paraplegia or Quadriplegia
- Tendon reflexes: Brisk
- Plantar reflex: Extensor
- Ataxia
- Dystonia
- Muscle: Atrophy
- Thyroid dysfunction: No clinical signs
- Laboratory
- MRI, Brain: Delayed myelination; Leukodystrophy
- Thyroid: T4 reduced; T3 increased; TSH normal or increased
- Carrier females
- Common clinical: Mild thyroid phenotype; No Neurologic features
- Occasional: AHDS with non-random X-inactivation
- Pregnancy: Child may have neonatal hypothyroidism
- Genetics
- Spastic paraplegia: Mental retardation & Strabismus
13
● Chromosome X; Recessive- Spastic paraparesis: Lower extremities; Slowly progressive
- Mental retardation: Severe
- Cranial nerves: Strabismus; Facial hypotonia
- Skeletal: ? Short & thick distal phalanges in some patients
- Woods-Black-Norbury Syndrome
● Chromosome Xq26-qter; Dominant- Females
- Males with congenital hypotonia & early death
- Rett syndrome
32
● Methyl-CpG-Binding protein 2 (MECP2); Chromosome Xq28; Dominant (or Sporadic) with male lethality
- Epidemiology
- Frequency: 1/15,000 females
- Sex distribution
- Females: Typical clinical syndrome
- Hemizygous males: Lethal or Neonatal encephalopathy
- Genetics
- Most mutations occur de novo
- Mutations: Missense, Nonsense, Frameshift
- C to T transition mutations: 70% at eight different CpG dinucleotides in MECP2 gene
- Non-affected females with mutation have skewed X-inactivation
- MECP2 mutation frequency
- Sporadic cases: 70% to 90%
- Familial cases: 50%
- Variant forms: 20% to 40%; More common with preserved speech variant
- Clinical-Genetic correlations
- X chromosome inactivation: Explains some clinical variabiltiy
- Inactivation of mutated chromosome: Unaffected carrier females
- Inactivation of normal chromosome: Severe disorder
- Severe phenotype: Truncation mutations early in gene
- Speech preserved: Missense or late truncation mutations
- Males
- Correlation with mutation type is especially clear
- Missense mutations with nonspecific X-linked retardation syndromes
- X chromosome inactivation: Explains some clinical variabiltiy
- Allelic disorders
- Nosology/Categorization: Epilepsy-dyskinesia syndromes
152
- Also see: ATP1A3, GNAO1
- Also see: ATP1A3, GNAO1
- Autism susceptibility, X-linked 3 (AUTSX3)
- Encephalopathy, neonatal severe
- Intellectual developmental disorder, X-linked syndromic 13 (MRXS13)
- Intellectual developmental disorder, X-linked syndromic, Lubs type (MRXSL)
- Rett syndrome, typical, atypical, or preserved speech
- Nosology/Categorization: Epilepsy-dyskinesia syndromes
152
- MECP2 protein
- Selectively binds CpG dinucleotides in DNA
- Mediates transcriptional repression
- Target genes: Retroviral, Tissue-specific (Leukosialin; Multi-drug resistance), Imprinted (Ube3a)
- Interactions: Histone deacetylases; Corepressor SIN3A complex; c-Ski; N-CoR
- Location: Higher levels in brain, especially neurons
- Clinical
- Course
- Normal development until 6 to 18 months
- Arrested development & decline over 10 to 15 years
- Mortality rate is 1.2% per year
- Deaths may be sudden & unexplained (25%)
- Cortical
- Mental retardation: Severe
- Cognitive: Absent language
- Seizures (50%)
- Grinding of teeth
- Loss of purposeful hand skills
- Other CNS
- Spastic paraparesis: Late
- Gait disorder: Ataxia/Apraxia
- Breathing patterns: Irregular, especially in waking state
- Autonomic
- Vasomotor disturbances: Legs
- Sialorrhea
- Constipation
- Skeletal
- Microcephaly
- Growth retardation
- Scoliosis or Kyphosis
- Cardiac: Long QTc interval
- Course
- Laboratory
- EEG: High amplitude spike & wave
- Brain: Low weight; Dendrites with reduced length & complexity
- Variant Rett (MECP2) syndromes
- Mild
- forme fruste: Worndown form
- Late regression
- Preserved speech
- Severe
- Congenital
- Encephalopathy
- Early seizure onset
- Males
- Genetics: Hemizygous MECP2 mutations
- Typical clinical
- Neonatal encephalopathy
- Death: 1¢2 years; May be prenatal
- Occasional patients: Typical Rett syndrome
- Associated with somatic mosaicism or partial or complete Klinefelter (47,XXY) karyotype
- Mental retardation with psychosis, Pyramidal signs & Macroorchidism (MRXS13)
● MECP2; Chromosome Xq28; Recessive
- Genetics: Allelic with Rett syndrome
- Clinical
- Mental retardation
- Manic-depressive episodes
- Increased muscle tone
- Hyperactive tendon reflexes
- Tremor
- Shuffling gait
- Macroorchidism
- Spasticity, Progressive & Mental retardation
14
● MECP2; Chromosome Xq28; Recessive
- Genetics
- Allelic with Rett syndrome
- Truncation mutation C1216T in exon 3
- Clinical
- Sex distribution: Males
- Cortical
- Cognitive: Absent language
- Seizures
- Grinding of teeth
- Motor
- Delayed development: Walking at 2 to 5 years
- Spastic paraparesis
- Facial hypotonia
- Ataxia: Gait
- Autonomic
- Sialorrhea
- Diarrhea
- Head circumference: Larger than average
- Genetics
- Mild
- Epidemiology
- Infantile spasms, Mental retardation & Spasticity (Epileptic Encphalopathy, Early-onset; EIEE1)
● Aristaless-related homeobox (ARX); Chromosome Xp21.3; Recessive
- Genetics: Mutations
- Types
- Polyalanine repeats
- Missense
- Deletion
- Clinical correlations
- Malformation phenotypes: Associated with protein truncation mutations & missense mutations in homeobox
- Non-malformation phenotypes: Missense mutations outside of homeobox & expansion of PolyA tracts
- Longer repeat mutations: Early Infantile Epileptic Encephalopathy With Suppression-Burst Pattern (Ohtahara Syndrome)
- Types
- Allelic disorder: X-linked infantile spasm syndrome (ISSX1; West syndrome)
- Genetics: Mutations
- Onset: Neonatal
- Clinical: Varied manifestations
- Mental retardation (100%): Most often moderate to severe
- Seizures (58%): Especially infantile spasms
- Hypotonia
- Spasticity
- Ataxia
- Dystonia: Episodic
- Macro- or Microcephaly
- Variant syndromes
- X-linked myoclonic epilepsy with spasticity and intellectual disability
- Infantile spasms with severe dyskinetic quadriparesis: Expansion of the first PolyA tract
● Chromosome Xq24-q25; Recessive
- Epidemiology: Brazilian family, 12 affected members
- Genetics
- Different locus from initial publication
- Clinical
- Onset
- Age: 10 to 25 years
- Gait: Shuffling
- Spastic paraplegia
- Predominantly legs
- Reflexes
- Tendon: Brisk
- Plantar: Extensor
- Clonus: Ankles
- Sensation
- Vibration: Reduced in legs after 6th decade
- Pain: Spontaneous; Legs
- Normal: Bladder & Bowel; Mental
- Course
- Progressive
- Wheelchair in 3rd or 4th decades
- Onset
● ATPase, H+ transporting, lysosomal, accessory protein 2 (ATP6AP2)
- Epidemiology: 1 Danish & German American family
- Genetics
- Mutation: Synonymous variant; c.345C>T (p.S115S); Markedly increases exon 4 skipping
- Allelic disorders
- X-linked mental retardation Hedera type (MRXSH)
- Congenital Disorder of Glycosylation, Type IIr (CDG2R)
- X-linked mental retardation Hedera type (MRXSH)
- ATP6AP2 protein
- Clinical
- Onset ages: 14 to 58 years; Mean 39; Variable
- Spasticity (60%)
- May be onset symptom
- Paraparesis
- Tendon reflexes: Increased in 50%
- Plantar response: Extensor in 50%
- Parkinsonism (100%)
- Resting tremor
- Masked facies
- Bradykinesia
- L-dopa responsive
- Seizures: 1 patient
- Course: Slowly progressive
- Laboratory
- MRI: Diffuse atrophy; Ventricular enlargement
- CNS pathology: Aβ depositis; tau (4R)+ & GFAP+ processes
● CD99 antigen-like 2 (CD99L2)
- Epidemiology: 25 patients
- Genetics
- Mutations: Truncation
- Allelic disorder: Females
- CD99L2 protein
- Ubiquitinated
- Activating interactor of CAPN1
- Cleaved by calpain
- Synapse-related
- Clinical (Males)
- Onset age: Mean 43 years
- Spasticity (88%)
- Dysarthria (84%)
- Pyramidal signs (54%)
- Leg predominant
- Gait disorder
- Bladder dysfunction (52%)
- Dysphagia (40%)
- Ataxia (72%)
- Oculomotor disorders (58%)
- Sensory loss (54%)
- Laboratory
- Brain MRI: Often normal
- CD99L2 variant: Spasticity or Ataxia, X-linked, Females
- Epidemiology: 4 patients
- Genetics
- Inheritance: Semi-Dominant
- Clinical: Partial presentations
- Onset age: 68 years
- Patients may have some of
- Spasticity
- Ataxia
- Sensory loss
- Oculomotor
- Bladder dysfunction
- Dysphagia
● Recessive
- Onset: ~10 years
- Clinical
- CNS
- Spastic paraparesis
- Tremor
- Deafness
- Cataracts
- Short stature
- Endocrine: Hypogonadism
- CNS
- Lab: Normal very-long-chain fatty acids
● Arginase, liver (ARG1)
- Genetics
- Mutations: Missense; Deletion; Stop
- Clinical
- General
- Growth failure
- Anorexia
- Vomiting
- Neurologic
- Spastic quadriplegia: Progressive
- Seizures
- Developmental delay
- Mental retardation
- Behavioral: Hyperactivity; Irritability
- General
- Laboratory
- Hyperammonemia
- Hyperarginemia
- Diaminoaciduria (Arginuria, Lysinuria, Cystinuria, Ornithinuria)
- Orotic aciduria
- Pyrimidinuria
- CSF amino acids: High
Syringomyelia
|
Familial syringomyelia: with Arnold-Chiari & Scoliosis
● ? Autosomal Dominant Cleidocranial dysplasia ● Runt-related transcription factor 2 (RUNX2)
● Growth/differentiation factor 5 (GDF5)
|
Cleidocranial dysplasia
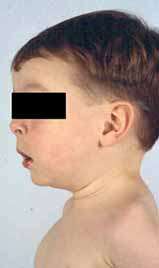  from A Kornberg MD |
Familial Spine Disorders & Myelopathy
|
Achondroplasia: FGFR-3; 4p16 Ankylosing spondylitis: 6p21 Atlanto-Axial instability Cleidocranial dysplasia: RUNX2; 6p21 Coffin-Lowry: RPS6KA3; Xp22 Collagen, type II disorders Disk disease: Multiple loci Dyggve-Melchior-Clausen: FLJ90130; 18q21 Hypophosphatemia; Rickets Larsen: Filamin B; 3p14 Lumbar stenosis Morquio Oculodentodigital Dysplasia: GJA1; 6q22 Opsismodysplasia: INPPL1; 11q13 Posterior longitudinal ligament Ossification Thickening Pseudoachondroplastic dysplasia: COMP; 19p13 Scheuermann disease Spondyloepiphyseal dysplasia variant Spondylometaepiphyseal dysplasia: DDR2; 1q23 Spina bifida Spinal Stenosis Tethered cord Trisomy 21 |
Also see: Acquired systemic causes of spine disorders Aggrecan mouse Radiology: Spine; Cervical spine 
|
Spine disorders, Familial
- Coffin-Lowry Syndrome
● Ribosomal Protein S6 kinase, 90 kD, Polypeptide 3;
Xp22.12; Dominant
- Genetics: Multiple mutation types
- Missense; Nonsense mutations; Splicing errors; Deletions or Insertions, Short
- RSK6 protein: Growth factor-regulated serine/threonine kinase
- Clinical features
- Cervical myelopathy: Due to hypertrophy ± calcification of ligamentum flavum
- CNS: Mental retardation; Deafness; Drop episodes
- Visceral neuropathy
- Skeletal
- Short, hyperextensible fingers & Flat feet
- Short stature
- Face: Pugilistic nose, Thick lips, Hypertelorism, Broad ears
- Systemic
- Mitral regurgitation
- Hydronephrosis
- Emphysema
- Female heterozygotes: Mild expression
- Learning disability
- Dysmorphism: Facial & Digital; Mild
- Laboratory
- Single-membrane-limited inclusions in fibroblasts
- Proteodermatan sulfate storage
- External link & Image: Coffin-Lowry Syndrome Foundation
- Genetics: Multiple mutation types
- Morquio's Syndrome
- Type A
● Galactosamine sulfatase; Chromosome 16q24.3; Recessive
- Type B
● β-galactosidase deficiency;
Chromosome 3p22.3; Recessive
- Clinical features
- Cervical myelopathy: Due to atlanto-axial dislocation
- Corneal opacities
- Hearing loss
- Joint laxity
- Growth retardation
- Hip dysplasia
- Type A
- Achondroplasia
● Fibroblast growth factor receptor-3;
Chromosome 4p16.3; Dominant
- Genetic
- Homozygotes with more severe disease
- Most cases new mutations (> 75%)
- Clinical features
- Myelopathy: Cervical & lumbar; Due to spinal stenosis
- CNS: Occasional hydrocephalus
- Skeletal
- Short stature
- Large head
- Brachydactyly
- Hearing loss
- Respiratory insufficiency
- Obesity
- Contractures
- Genetic
- Type II Collagenopathies (Cartilage & Vitreous collagen)
2
● Collagen type II, α-1 chain (COL2A1); Chromosome 12q13.11; Dominant
- Achondrogenesis II
: Lethal
- Hypochondrogenesis
: C1-C2 dyslocation
- Spondyloepiphyseal dysplasia (SED) congenita
: C1-C2 dyslocation
- Spondyolo-epi-metaphyseal dysplasia (SEMD) Strudwick
: C1-C2 dyslocation
- Kneist dysplasia
: C1-C2 dyslocation
- Stickler dysplasia
: Spinal stenosis
- Other Stickler syndrome dominant mutations: COL11A1
;
COL11A2
- Stickler syndrome recessive mutations: COL9A1
;
COL9A2
- Other Stickler syndrome dominant mutations: COL11A1
- Type II collagen absent in herniated human intervertebral disc samples
- Achondrogenesis II
- Spondyloepiphyseal dysplasia (SED) with atlantoaxial instability
:
C1-C2 dyslocation
● Autosomal Dominant- Cervical myelopathy due to atlanto-axial dislocation
- Spondylometaepiphyseal dysplasia, Short limb-hand type
● Discoidin domain receptor family, member 2 (DDR2); Chromosome 1q23.3; Recessive- Cervical myelopathy due to atlanto-axial dislocation
- Bone dysplasia: Severe short-limb
- Short stature
- Face: Short nose; Ocular hypertelorism; Retro/micrognathia
- Chest: Narrow; Abnormal sternum; Short ribs
- Pseudoachondroplastic dysplasia
● Cartilage oligomeric matrix protein;
Chromosome 19p13.11; Dominant
- Dwarfism: Short-limb
- Limbs: Brachydactyly; Telescoping fingers
- Joints: Limited elbow and hip extension; Ligamentous laxity
- Cervical myelopathy due to atlanto-axial dislocation
- Recessive type pedigrees due to gonadal mosaicism
- Cytoplasmic metachromasia of fibroblasts
- Dyggve-Melchior-Clausen disease
34
● Dymeclin (DYM; FLJ90130)
; Chromosome 18q21.1; Recessive
- Genetics
- Allelic with: Smith-McCort dysplasia (SMC)
- Allelic with: Smith-McCort dysplasia (SMC)
- Clinical
- Skeletal
- Dwarfism: Short trunk
- Head: Prominent jaw; Microcephaly
- Limbs: Claw hand; Restricted joint mobility; Hip subluxation
- Chest: Barrel; Protuding sternum
- Spine: Osteochondrodysplasia
- Cervical myelopathy: Due to atlanto-axial dislocation
- CNS: Mental retardation
- Skeletal
- Laboratory
- Mucopolysacchariduria
- Abnormal cartilage (Growth plate) histology
- Disordered chondrocyte columns: Degenerating cells; Rough ER inclusions
- Brain: Hypomyelination
- Genetics
- Larsen syndrome (Osteochondrodysplasia)
45
● Filamin B; Chromosome 3p14.3;Dominant
- Filamin B disorders
- Perinatal lethal atelosteogenesis I
& III
- Spondylocarpotarsal syndrome
- Perinatal lethal atelosteogenesis I
- Genetics: Missense mutations
- Filamin B protein
- Filamins: Cytoplasmic; Crosslink actin into 3D networks
- Filamin B: Growth plate chondrocytes; Developing vertebral bodies
- Clinical
- Short stature
- Face: Flat; Frontal bossing; Hypertelorism
- EENT: Cataract; Cleft-palate
- Spine & Skeletal
- Scoliosis
- Abnormal vertebral segmentation & development: Kyphosis
- Joint dislocations: Knee
- Arthrogryposis
- Limbs
- Multiple congenital joint dislocations
- Brachydactyly
- Carpal or phalangeal abnormalities
- GU: Cryptorchidism; Genital anomalies
- CNS
- Myelopathy
- Cervical spine
- 2° to abnormal vertebral segmentation: Hypoplastic C4 or C5
- Infantile onset
- Hydrocephalus
- Myelopathy
- Variant: Larsen syndrome, Recessive
● β-1,3-Glucuronyltransferase 3 (B3GAT3); Chromosome 11q12.3; Recessive
- Filamin B disorders
- Opsismodysplasia
● Inositol Polyphosphate Phosphatase-like 1 (INPPL1); Chromosome 11q13.4; Recessive
- General classification: Severe spondylodysplastic dysplasias (group 14)
- INPPL1 protein
- Inositol-1,4,5-trisphosphate 5-phosphatase family
- Negative regulation of insulin signaling & glucose homeostasis
- Docking protein
- Endochondral ossification
- Polymorphisms associated with predisposition to insulin resistance
- Clinical
- Cervical myelopathy due to atlanto-axial dislocation
- Hypotonia
- Craniofacial: Prominent brow, Depressed nasal bridge, Small anteverted nose, Long philtrum
- Death
- Respiratory failure in 1st few years in some
- Some long-term survivors
- Skeletal dysplasia: Late bone maturation
- Dwarfism
- Short hands & Feet
- Radiology
- Platyspondyly
- Metacarpals: Square
- Skeletal ossification: Delayed
- Metaphyseal cupping
- Spina bifida
- Associated with
- Anencephaly
- Hydrocephalus
- Lipoma
- Sacral: Hairy patch, dimple, agenesis (caudal regression)
- Meningocele
- Posterior
- Anterior: Autosomal dominant
- Onset: Symptoms in young adults with obstructive labor
- GI: Constipation & rectal fistula & abscess
- Meningitis
- Onset: Symptoms in young adults with obstructive labor
- Posterior
- Clinical
- Weakness: Legs
- Sensory loss: Sacral
- Urinary incontinence
- Mouse model: Double heterozygotes for undulated
& patch
- Rule out: Sacral dysgenesis or hemisacrum
-
● X-linked Dominant
● Autosomal Dominant
- Associated with
- Trisomy 21
- Cervical myelopathy due to atlanto-axial instability
- Familial intervertebral disc disease
- Intervertebral disc disease: General
67
- Prevalence of degenerative intervertebral discs increases linearly with age
- 80% of all lumbar discs abnormal at 70 years of age
- 43.8% of patients with lumbar disc herniation have family history in patients younger than 17 years old
- Lumbar disc herniation in juvenile patients 5x greater with positive family history
- Family history associated with more severe disc disease
- Similar familial predisposition for cervical & lumbar disc disease
- Monozygotic twins
- Heritability of disc height & bulge = 60% to 74%
- Association between lumbar disc degeneration on MRI & propensity to report pain in lumbar spine
- SPG25: Disc herniation & Spastic paraplegia
27
● Chromosome 6q23.3-q24.1; Recessive- Epidemiology: Family from Northeastern Italy
- Clinical
- Onset age: 30 to 46 years
- Spine pain
- Early in disease course
- Cervical & Lumbar
- Radiates to extremities
- Spastic paraparesis: Legs
- Disc herniations: Cervical, Thoracic & Lumbar
- Neuropathy: Mild; Sensory or Motor
- Treatment: Surgical
- Radiology
- Disk herniations
- Intervertebral disc disease: Increased risk
● Collagen IX α2 (COL9A2);
Chromosome 1p34.2; Dominant
- Mutation: Gln326Trp
- Allelic COL9A2 disorders
- Multiple epiphyseal dysplasia, Type2
- Stickler syndrome, type V
- Epiphyseal dysplasia + Myopathy
- Multiple epiphyseal dysplasia, Type2
- Intervertebral disc disease: Increased risk
● Collagen IX α3 (COL9A3);
Chromosome 20q13.33; Dominant
- Mutation: Arg103Trp
- Present in 12.2% of lumbar disc disease cases and in 4.7% of controls
- Increased the risk of lumbar disc disease about 3-fold
- Allelic COL9A3 disorders
- Multiple epiphyseal dysplasia, Type3
;
- Epiphyseal dysplasia + Myopathy
- Multiple epiphyseal dysplasia, Type3
- Mutation: Arg103Trp
- Intervertebral disc disease: Increased risk
● Cartilage intermediate layer protein (CLIP);
Chromosome 15q22.31; Dominant
- CLIP protein
- Expressed abundantly in intervertebral discs
- Colocalizes with TGFB1
in clustering chondrocytes & territorial matrices
- Inhibits TGF-β1-mediated induction of cartilage matrix genes
- Expression increases as lumbar disc degeneration progresses
- Mutation: Ile395Thr
- Increased frequency with lumbar disc disease
- CLIP protein
- Intervertebral disc disease, lumbar: Increased risk in Japanese population
66
● Thrombospondin II (THBS2);
Chromosome 6q27
● Matrix metalloproteinase-9 (MMP9);
Chromosome 20q13.12
- THBS2
- Mutation: Intronic SNP (IVS10-8C to T)
- Present in Japanese populations
- Located in a polypyrimidine tract upstream of 30 splice site of intron 10
- Exerts allelic differences on exon 11 skipping rates in vivo
- Susceptibility allele shows increased skipping
- Skipping of exon 11 results in: Decreased THBS2 interaction with MMP2 and MMP9
- Increased the risk of lumbar disc disease: ~1.4-fold
- THBS2 protein
- High levels in intervertebral discs
- Interacts with MMP2 & MMP9 (Gelatinases)
- Promotes LRP
-mediated endocytosis of MMP2 & subsequent lysosomal degradation
- Thrombospondin family
- Family of five secreted, modular glycoproteins
- Location: Extracellular matrix
- Mutation: Intronic SNP (IVS10-8C to T)
- MMP9
- MMP9 Mutation
- Missense: Q279R missense
- Increased risk: ~1.3-fold
- MMP9 mutation also associated with: Lung cancer metastasis & Trachoma
- MMP9 Mutation
- MMP9 protein
- THBS2
- Combined risk for lumbar disc herniation of THBS2 + MMP9 mutations: ~3-fold
- Intervertebral disc disease: General
67
- Lumbar disc disease: Association with severe degeneration
53
● Aggrecan; Chromosome 15q26.1
- Mutation: Polymorphism
- Exon 12 is candidate marker for severe lumbar disc disease
- Shorter number of repeats with multilevel and severe disc degeneration
- Disorders associated with severe disc degeneration
- Collagen II
● Col2A1; Chromosome 12q13.11-q13.2
- Spondyloepiphyseal dysplasia
- Achondrogenesis
- Stickler syndrome
- Spondyloepiphyseal dysplasia
- Cartilage oligomeric matrix protein
● COMP; Chromosome 19p13.1
- Pseudoachondroplasia
- Multiple epiphyseal dysplasia, Fairbank type
- Pseudoachondroplasia
- Collagen II
 Ankylosing Spondylitis Limited back flexion |
● Chromosome 6p21.3; Dominant
- Onset
- 15 to 30 years
- Back pain
- Large joints
- Epidemiology
- Male predominant: 75%
- Prevalence: 1% to 2% of population
- Clinical
- Cervical myelopathy
- 86% Male
- 2° Atlanto-axial instability: 20%
- Disease duration > 10 years
- Skeletal
- Back
- Pain: Variable
- May radiate to legs 2° cauda equina fibrosis
- Better with activity
- Worse with rest
- Worse in AM
- Tenderness over sacroiliac joints
- Stiffness: Limited flexion & amp; extension
- Progression: Upward from lumbar spine
- Pain: Variable
- Chest wall
- Limited movement
- Costochondral joint involvement
- Joint arthritis: Transient 50%; Permanent 25%
- Back
- Systemic features
- Cardiac (3% to 5%): AV block; Aortic insufficiency
- Pulmonary fibrosis: Late in disease
- Iridocyclitis: 20%
- Amyloidosis
- Progression
- 10% Disability in 10 years
- Worse prognosis: Hip disease in 1st 2 years
- Cervical myelopathy
- Laboratory
- HLA-B27 haplotype association: 90%
- Erythrocyte Sedementation Rate: High
- X-ray: Earliest changes in sacroiliac joints; Bamboo spine
- Anemia: Mild
- Radiology: Changes are Bilateral & Symmetric
- Pathology
- Inflammation at sites of insertion of ligaments & tendons in bones
- Reactive bone growth
- Treatment: Exercise; NSAIDs
- External links
- eMedicine
- MedMedia
- Images: Sacroiliac joints; Facet joints
● Connexin 43 (GJA1)
- Allelic disorders
- GJA1 protein
- Clinical
- Dysmorphisms
- Craniofacial
- Ocular: Microphthalmia, Microcornea, Iris abnormalities, Cataracts, Glaucoma, Optic atrophy
- Nasal
- Dental
- Limb
- Syndactyly type III: Occasional
- Craniofacial
- CNS
- Spastic paraplegia
- Onset age: 2nd decade
- Gait disorder
- Spastic bladder
- Mental retardation: Mild
- MRI: Leukodystrophy in subcortical cerebral white matter
- Course: Slowly progressive
- Spastic paraplegia
- Conductive deafness: Occasional
- Cardiac abnormalities: Rare
- Dysmorphisms
● Multiple susceptibilty loci; Recessive
|
|
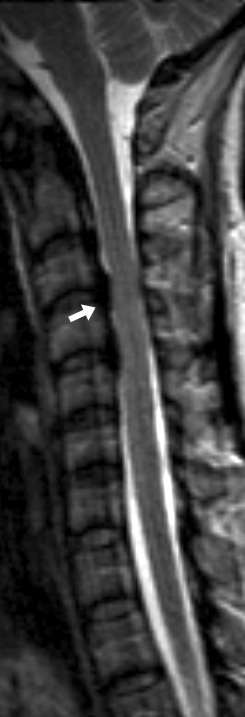
{kind=link}
{kind=link}
- Genetic associations
- HLXB9
- Currarino triad
:
Anal abnormalities, Anterior meningocele, Sacral abnormalities
- Currarino triad
- VATER & VACTERL syndromes
- CHARGE
- Chromosome 22: Deletion 22q11.2
; Ring chromosome
- DiGeorge syndrome: Clinical; No chromosome abnormality
- Chromosome 21: Trisomy 21
; Trisomy 21q + Monosomy of 6q
- Other chromosomes: Trisomy 8; Partial monosomy 4p15.2-pter + Partial trisomy 13q32-qter
- Other syndromes:
Klippel-Trenaunay-Weber
,
Klippel-Feil
, Dandy-Walker, Fuhrmann
- FG syndromes
- HLXB9
- Anatomic associations: Various spinal dysraphic states
- Spinal lipomas
- Diastematomyelia
- Dermal sinus
- Myelocystocele
- Tight filum terminale
- Loosely defined "tethered cord"
- Onset of signs
- Most common in children or young adults
- May occur at any age
- May be associated with exercise or growth
- Weakness
- Legs
- May be asymmetric
- Pain
- Lower back
- Radiation: To the legs, perineum, or genitals
- Flexion of the back: Increased pain
- Lordotic posture or pelvic tilting: Decreased pain; Worse gait
- Other neurologic
- Gait disorders: Associated with weakness or spasticity
- Urinary incontinence: Failure to toilet train
- Sensory loss: Early in perineal region
- Tendon reflexes: May be reduced or increased
- Scoliosis
- Cutaneous changes: May be prominent or absent
- Hairy patch: Commonly associated with diastematomyelia
- Nevus
- Appendage or skin tag
- Small dimple with pinhole: Especially at lumbosacral junction; Associated with dermal sinus
- Large subcutaneous fat collection: Associated with spinal lipomas
- Treatment: Surgery
- Most likely to improve: Pain
- Worst prognosis: Bladder function
- Early: Dwarfism, mild
- Age-related: Lordosis; Spastic paraparesis
- Pathology
- Herniation & degeneration of vertebral disks
- Degeneration of disc chondrocytes
|
Arnold-Chiari Malformation ● Autosomal Recessive or Sporadic ● With Syringomyelia
|
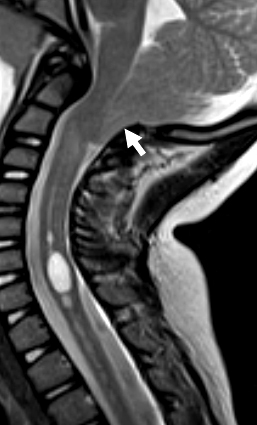 Arnold-Chiari malformation + Hydromyelia
Cerebellar vermis projects below foramen magnum (Arrow) |
 From: Simon Kornberg Canine ACM + Hydromyelia
|
{kind=link}
Spastic Ataxia Syndromes
87
|
Hereditary Dominant SCA SPAX Recessive SPAX SPG X-linked Acquired |
Spastic Ataxia, Dominant
|
Spastic Ataxia, Recessive
|
|
Spastic Ataxia: X-linked
Spastic Ataxia: Acquired
|
Spastic Ataxia 1, Hereditary (SPAX1)
● Vesicle-related membrane protein 1 (VAMP1; Synaptobrevin; SYB1)
- Epidemiology: Newfoundland families
- Genetics
- Mutation: Splice site, c.340+2T>G; Ser114Arg
- Mutation leads to: Loss of VAMP1A isoform
- Allelic disorder: Congenital Myasthenic Syndrome, Presynaptic
- VAMP1 protein
- General: Synaptic vesicle cycle at Presynaptic nerve terminals
- Family: VAMP/Synaptobrevin
- Location: Post-Golgi vesicular compartments
- Anchored in vesicle membrane by: C-terminal domain
- Interact with: Synaptophysin prior to docking of vesicle
- After docking: Bind to SNAP-25-syntaxin complex
- Forms: Synaptic core complex
- Exocytosis, Regulated
- Starts cascade of protein interactions leading to: Neurotransmitter exocytosis
- Cleaved by some botulinum toxins
- Clinical
- Severity variable within families
- Onset
- Age
- Variable
- Typical: 10 to 20 years
- Range: Early childhood to 20's
- No anticipation
- Leg spasticity
- Age
- Spasticity
- Legs
- Severe
- Tendon reflexes: Brisk
- Cerebellar
- Involuntary head jerks
- Dysarthria
- Dysphagia
- Gait disorder
- Ocular: Supranuclear gaze palsy
- Sensory: Vibration loss in legs
- Not affected: Life span; Cognition
- Pathology
- Degeneration of: Corticospinal tracts & Posterior columns
- VAMP1 variant disorder: Congenital Myasthenic Syndrome, Presynaptic 25 (CMS 25)
116
- Epidemiology: 11 families
- Genetics
- Inheritance: Recessive
- Mutations: Frameshift or Missense; Most loss of function
- Allelic disorder: SPAX1, Dominant
- Clinical
- Onset age: Birth
- Early motor: Hypotonia; Feeding difficulties; Developmental delay
- Weakness: Diffuse; bulbar; Respiratory
- GI: Dysphagia; Aspiration with chest infections
- Eye: Ophthalmoparesis; Strabismus
- Joints: Contractures or Laxity
- Pyridostigmine treatment: Mild improvement
- Laboratory
- Serum CK: Normal
- Muscle biopsy: Myopathic; Atrophy, especially type 2; Hypertrophy, Type 1
- EMG: Myopathic
- NCV: CMAP small amplitude
- RNS: 2x Increment at 20 Hz or Normal
- Leg MRI: Mild change in adductors & quadriceps
Hereditary Spastic Ataxia (SPAX7)
● Autosomal Dominant
- Epidemiology: 3 families
- Clnical
- Onset age: Congenital to 18 years
- Eye
- Optic atrophy
- Pupils: Miosis; Reduced reactivity to light
- Ptosis
- Ataxia: Limbs; Gait; Dysarthria
- Pyramidal
- Spastic paraparesis
- Tendon reflexes: Brisk in legs
- Plantar response: Extensor or Flexor
- Extrapyramidal: Chorea
Spastic Ataxia 2 (SPAX2; SPG 58)
● Kinesin family member 1C (KIF1C)
- Nosology: SPG58
- Epidemiology: Moroccan, Algerian, Palestinian & French families
- Genetics
- Mutations: Gly102Ala; Arg731*; Arg169Trp; Pro176Leu; G183A (Splice); rg301Gly; R731X (Stop); Deletion
- Allelic disorder
- KIF1C heterozygote: Mild syndrome
- KIF1C protein
- Axon transport, Anterograde
- Kinesins: Other disorders
- Clinical
- Onset
- Age: Range 1 to 69 years; Often 6 to 18 years
- Dysphagia
- Falling
- Cerebellar ataxia
- Dysarthria
- Gait disorder
- Nystagmus: Horizontal; Some patients
- Tremor & Head titubation
- Pyramidal
- Spasticity: 1 of 3 families; All limbs
- Tendon reflexes: Brisk in legs
- Plantar response: Extensor
- Arms: Often normal
- Weakness: Some patients
- Distal
- Hands & Feet
- Fasciculations
- Sensation: Normal or Reduced (Panmodal; Posterior column)
- Bladder: Often normal
- Progression
- Over few years or Slow
- Often still walking after 2 decades
- EOM: Saccadic pursuit; Dysmetric saccades; Upgaze paresis (1 patient)
- Cognition: Intact
- Onset
- Laboratory
- Brain MRI: Normal or Internal capsule demyelination; T2 hyperintensities
- VEP: Normal or Increased latency
- NCV: Demyelination in some
- KIF1C heterozygotes
- Genetics: Semidominant inheritance
- Clinical
- Weakness: Hip abductors; Proximal legs
- UMN: Tendon reflexes brisk; Extensor plantar responses
- NCV: Neuropathy
Branchial Myoclonus with Spastic paraparesis & Cerebellar ataxia
● Glial fibrillary acidic protein (GFAP)
- Nosology: Adult onset Alexander disease
24
- Genetics
- Mutation: Val87Gly; Missense
- GFAP protein
- Intermediate-filament (IF) protein
- Location: Cytoplasm
- Specific for cells
- Astroglial lineage
- Lacking fibronectin
- GFAP immune disorder: Astrocytopathy
- Clinical
- Onset age: 12 to 71 years
- Branchial (Palatal) myoclonus
- Cerebellar
- Truncal ataxia
- Nystagmus
- Bulbar: Dysarthria; Dysphagia; Dysphonia
- Spastic paraparesis (50%)
- Reflexes
- Tendon: Mildly increased
- Plantar: Extensor
- Autonomic: Bladder dysfunction in some
- Cognitive: Normal
- Progression: Death or severe disability 5 to 10 years after onset
- Pathology (MRI)
- Medulla & Spinal cord: Severe atrophy
- Cerebral & Cerebellar cortex: Mild atrophy
- Periventricular: Intensity on T2
- Inferior olive: Hypertrophy (30%)
- GFAP immune disorder: Astrocytopathy
138
- Clinical
- Onset
- Antecedent: Flu-like symptoms (90%)
- Course: Subacute
- Ages: 28 to 72 years; Mean 53 years
- Polyradiculopathy (10% to 20%)
- Meningoencephalomyelitis
- Pyramidal signs
- Brainstem
- Tremor
- Psychiatric Δ
- Neoplasm association: Ovary, Teratoma, CNS lymphoma
- Course: Acute monophasic or Chronic
- Treatment: Immunothereapy, corticosteroid or other
- Onset
- Laboratory
- MRI: Linear gadolinium meningeal enhancement
- CSF: Pleocytosis
- GFAP Antibody
- Cell target: Astrocytes
- Antigen target: GFAPα
- May be present without CNS astrocytopathy
- Other antibodies: NMDAR in 20%
- Neuropathology: CD8 lymphocytes
- Other: Antibody related CNS syndromes
- Clinical
Spastic ataxia, congenital, with Mental retardation & Osmiophilic skin vessels (GAMOS1; SCAR5)
● WDR73
- Nosology: Galloway-Mowat Syndrome 1
- Epidemiology: > 50 patients
- Genetics
- Mutation: H347Y; Homozygous
- Initially misidentified as ZNF592
mutation
- WDR73 protein
- Clinical
- Onset: Congenital
- Ataxia
- Spasticity: Brisk tendon reflexes; Speech disorder
- Strength: Mildly reduced in legs
- Gait: 60% never walk
- Mental retardation: Severe; Little speech
- Eye: Optic atrophy; Oculomotor apraxia
- Sensory: Normal
- Skeletal: Microcephaly; Short stature
- Course: Non-progressive
- Laboratory
- Skin vessels: Osmophilic endothelium
- MRI: Cerebellar atrophy
- Muscle biopsy: Normal morphology & mitochondrial studies
Neurodegeneration with Optic atrophy, Childhood-onset (NDGOA)
● Ubiquitin carboxyl-terminal esterase L1 (UCHL1; PGP 9.5)
- Epidemiology: Turkish family
- Genetics
- Mutation: Homozygous; Missense; Glu7Ala
- UCHL1 protein
- Clinical
- Onset
- Age: Childhood
- Visual loss at 5 years
- Early development: Normal
- Eye: Optic atrophy, Nystagmus, Blindness
- Cerebellar ataxia: Inability to stand
- Sensory loss: Vibration; Joint position
- Spasticity
- Legs
- Tendon reflexes: Increased
- Plantar responses: Extensor
- IQ: Mildly decreased
- Seizures: 1 patient
- Onset
- Laboratory
- EEG: 3.5- to 4-Hz spike activity
- NCV: Normal
- EMG: Myokymia or Myotonia
- Brain MRI: Cerebral & Cerebellar atrophy
Spastic Paraplegia, Ataxia ± Mental Retardation (SPAR)
● Autosomal Dominant
- Epidemiology: Michigan family
- Onset: 15 to 35 years; ? Anticipation
- Clinical: Variable phenotype
- Spastic paraplegia (100%)
- Leg spasticity
- Bladder dysfunction
- Dorsal column sensory loss: Reduced vibration & proprioception
- Tendon reflexes: Brisk in legs
- Plantar responses: Extensor
- Course: Progressive; Disabling
- Ataxia (65%)
- Limb
- Dysarthria
- EOM: Saccadic intrusions; Hypermetric saccades
- Dystonia (60%): Mild; Hands & Feet
- Mental retardation (30%): Mild; Non-progressive
- Spastic paraplegia (100%)
- Laboratory
- MRI: Spinal cord ± Cerebellar atrophy
- NCV & EMG: Normal
- SSEP: Conduction delay between spinal cord & cortex
- See: Spastic ataxias
Spastic Paraplegia, Epilepsy & Mental Retardation (SPERM)
● Autosomal Dominant
- Epidemiology: 1 Italian family
- Genetics: Incomplete penetrance
- Onset: < 40 years
- Clinical
- Motor (83%)
- Distribution: Lower limb involvement
- Weakness
- Spastic paraplegia
- Progressive defecit
- Epilepsy (100%)
- Mental retardation: Variable severity
- Motor (83%)
Lipodystrophy with Spastic-Ataxia & Congenital cataracts (LCCNS)
● Caveolin 1 (CAV1)
- Epidemiology: Kentucky family
- Genetics
- Mutation: Heterozygous; Truncating; 1- BP deletion
- Clinical: Variable neurologic phenotype
- Onset
- Age: Congenital
- Lipodystrophy
- Cataracts
- Lipodystrophy: Partial
- Absence of subcutaneous fat over entire body except buttocks, hips, and thighs
- Ocular
- Retinitis pigmentosa
- Iris deposits
- Spastic paraplegia
- Leg spasticity
- Bladder dysfunction
- Dorsal column sensory loss: Reduced vibration; Legs > Arms
- Tendon reflexes: Brisk in legs; Clonus
- Plantar responses: Extensor
- Course: Progressive & Disabling in 2nd & 3rd decade in some patients
- Cerebellar
- Spastic-Ataxic gait
- EOM: Gaze-evoked horizontal nystagmus; Upbeat nystagmus; Ocular dysmetria
- Orthostatic hypotension
- General: Neurologic involvement varied; Some patients with mild ataxia
- Onset
- Laboratory
- MRI: Spinal cord with increased T2 signal
- NCV & EMG: Normal
- SSEP: Conduction delay
- Lipids: High serum cholesterol & triglycerides
- Vitamin E & β-Tocopherol: Markedly elevated
- Carbohydrate metabolism: Insulin resistance; Impaired glucose tolerance
- Body fat
- Absent subcutaneous fat: Upper body & Trunk
- Preservation of subcutaneous fat: Between waistline & Knees
- Visceral fat accumulation
- Muscle biopsy: No mitochondrial changes
- See: Spastic ataxias
Paroxysmal Choreoathetosis/Spasticity (CSE; DYT9)
● SLC2A1 (GLUT1)
- Epidemiology: 4 families
- Genetics
- Mutations: Deletion, Stop & Missense
- Allelic disorders
- GLUT1 deficiency syndrome-1 (GLUT1DS1)
- GLUT1 deficiency syndrome-2 (GLUT1DS2)
- Idiopathic generalized epilepsy-12, susceptibility (EIG12)
- Dystonia 9
- GLUT1 deficiency syndrome-1 (GLUT1DS1)
- SLC2A1 protein
- Glucose & Dehydroascorbic acid transporter
- Clinical features
- Onset: 2 to 15 years
- Spasticity between episodes: 30%
- Episodes
- Involuntary movements & Dystonic postures
- Perioral & leg paraesthesias
- No ataxia
- Duration: 20 minutes
- Frequency: 2x/day to 2x/year
- Precipitated by: Exercise; Stress; Alcohol; Lack of sleep
- Treatment: ? Acetazolamide & Phenytoin
- Cognition: Impaired
- Laboratory
- EEG: Generalized slowing
- CSF Glucose & Lactate: Low
Hypomyelinating Leukodystrophy 6 (HLD6; HABC)
● Tubulin β4A (TUBB4A)
Hypomyelinating Leukodystrophy 9 (HLD9)
● Arginyl-tRNA Synthetase (RARS1)
- Epidemiology: 33 patients
- Genetics
- Mutations: Heterozygous; Missense, Splice, Stop; Loss of function
- RARS protein
- tRNA synthetase: Cytoplasmic
- Clinical: Spastic ataxia
- Onset age: Birth to 1 year; Mean 6 months
- Spasticity: Legs > Arms; Gait disorder; Pseudobulbar
- Cerebellar: Intention tremor, Dysmetria, Dysarthria, Nystagmus
- Cognition: Impaired; Language delay
- Course: Progressive
- Laboratory
- Brain MRI: Atrophy; Loss of myelin, supra- & infratentorial
Hypomyelinating Leukodystrophy (Spastic-Ataxia) 120
● Glutamyl-Prolyl-tRNA Synthetase (EPRS)
- Epidemiology: 5 patients, 4 families
- Genetics
- Mutations: Missense & Stop
- EPRS protein
- Amino-acyl-tRNA synthetase (ARS): Bifunctional
- Catalyzes aminoacylation of glutamic acid & proline tRNA
- Part of GAIT (γ-interferon-activated inhibitor of translation) comple
- Involved in selective translational silencing
- Components
- Ribosomal protein L13a (RPL13A)
- EPRS
- Synaptotagmin-binding cytoplasmic RNA-interacting protein (SYNCRIP, NSAP1)
- Glyceraldehyde 3-phosphate dehydrogenase (GAPDH)
- Ribosomal protein L13a (RPL13A)
- Clinical
- Onset age: Infant to 14 years
- Cognitive regression
- Spasticity
- Ataxia: Intention tremor; Dysarthria; Nystagmus
- Dystonia
- Dysphagia
- Optic atrophy: Visual loss
- Course: Progression to disability
- Laboratory
- Brain MRI: Hypomyelinating leukodystrophy; Thin corpus callosum
Leukoencephalopathy with Ataxia, UMN signs & Polyneuropathy (RPIAD)
● Ribose-5-Phosphate Isomerase (RPIA)
- Epidemiology: 4 patients
- Genetics
- Mutations: Missense & Stop
- RPIA protein
- Pentose phosphate pathway: Non-oxidative phase
- Converts ribulose-5-phosphate to ribose-5-phosphate
- Location: Cytosol & Membrane-bound organelles
- Deficiency: Leads to accumulation of pentoses & pentose phosphates
- Clinical
- Onset
- Infancy: 4 days to 6 months
- Psychomotor retardation: Slow walking & Speech
- Developmental & Speech delay
- Seizures: Onset 4 years
- Ataxia
- Arms & Legs
- Dysarthria
- Nystagmus: Lateral gaze
- Upper motor neuron
- Spasticity
- Hyperactive tendon reflexes: Except at ankles
- Plantar reflexes: Extensor
- Eyes: Optic atrophy; Retinitis pigmentosa
- Peripheral nerve
- Tendon reflexes at ankles: Reduced
- Motor: Mild distal atrophy & weakness
- Sensory loss: Distal
- Course: Progression after age 5 to 7
- Onset
- Laboratory
- Brain imaging
- MRI: Leukoencephalopathy; Cerebellar atropy
- Proton MRS: Ribitol & D-Arabitol peaks
- Ribitol & D-Arabitol high: Serum, Urine & CSF
- Lactate & Pyruvate: Normal
- Nerve conduction testing: Axon loss in some patients
- Motor: Mildly reduced velocities
- Sensory: Mildly reduced velocities
- Serum CK: Mildly high
- EEG: Slowing; Epileptic activity
- Brain imaging
Leukoencephalopathy with Cortical involvement & Peripheral neuropathy
● Folate receptor 1, adult (FOLR1)
- Nosology
- Neurodegeneration due to cerebral folate transport deficiency
- Cerebral folate deficiency
- Epidemiology: 13 families
- Genetics
- Mutations: c.466T>G (p.W156G); Gln118Ter; Cys175Ter; 18-BP Dup, NT130
- FOLR1 protein
- High affinity for folic acid & several reduced derivatives
- Mediates delivery of 5-methyltetrahydrofolate to cell interior
- Clinical
- Onset: Late infantile to 3 years
- Psychomotor regression, severe
- Hypotonia
- Epilepsy: Myoclonic
- Ataxia
- Spasticity
- Tendon reflexes: Increased or Decreased
- Polyneuropathy
- Treatment: Folinic acid
- Laboratory
- Brain MRI
- Cortical disturbances
- CNS hypomyelination
- Cerebellar atrophy
- NCV
- Axon loss: CMAPs small amplitude, some temporal dispersion
- NCV: 31 to 37 M/s
- CSF: 5-methyltetrahydrofolate low
- EEG: Multifocal epileptiform
- Brain MRI
Pelizaeus-Merzbacher¢Like Disease (PMLD; HLD2)
● Gap Junction Protein α12 (Connexin 46.6; Connexin 47; GJA12; GJC2)
- Nosology: Hypomyelinating leukodystrophy 2
- Epidemiology: Turkish & German families
- Genetics
- Missense mutation in one or both alleles; Some nonsense mutations
- Mild phenotype: Ile33Met
- Knockout mouse: Clinically normal
- Allelic with
- SPG 44
- Lymphedema, hereditary, IC
- GJA12 protein
- Gap junction protein
- Higher expression: Brain & Spinal cord
- Low expression: Skeletal muscle
- May play role in oligodendrocyte function
- Functional overlap with GJB1
- Clinical
- Onset
- Age: Early infancy
- Poor head & trunk control
- Nystagmus
- Motor development
- Impaired by age of 8 to 15 mo
- Delayed unaided sitting and/or walking
- Walking at age 5 rare
- Facial weakness: Common
- Cerebellar
- Ataxia
- Nystagmus
- Choreoathetosis
- Dysarthria
- Spasticity: Progressive
- Epilepsy: Focal; Onset prior to adolescence
- Peripheral nerve: Mild neuropathy
- Onset
- Laboratory
- Nerve conductions
- Motor & sensory velocities: Mildly slowed
- Lower limbs more involved
- MRI: Leukodystrophy
- Nerve conductions
- Variant syndrome (SPG44
):
Later onset complicated spastic paraplegia
73
- GJA12 (GJC2) Mutation: Ile33Met; Homozygous
- Mutant protein: Forms gap junction channels with altered functional properties
- Clinical
- Onset
- Age: 1st or 2nd decade
- Gait disorder
- Hand tremor
- Spastic paraplegia
- Legs > Arms
- Walking preserved: Until late
- Sphincter dysfunction: 1 patient late in course
- Sensory loss: 1 patient late in course
- Cerebellar ataxia: Mild; No nystagmus
- Mental impairment: Mild
- Progression: Slow
- Other
- Pes cavus
- Strabismus
- Scoliosis
- Onset
- Laboratory
- MRI: Hypomyelinating leukoencephalopathy
- Evoked potentials: Abnormal
- EMG & NCV: Normal
Pelizaeus-Merzbacher¢Like Disease 2
● Small inducible cytokine subfamily E, Member 1 (SCYE1; AIMP1)
- Nosology
- Disease name: Leukodystrophy, hypomyelinating, 3 (HLD3)
- SCYE1 protein: Aminoacyl tRNA synthase (ARS) complex-interacting multifunctional protein 1 (AIMP1); p43
- Epidemiology: Israeli Bedouin family
- Genetics: Frameshift mutation; Gln98ValfsX30
- AIMP1 protein
- Expression: Neurons of Brain & Spinal cord
- Associated with: Neurofilament light chain (NEFL)
- AIMP1 depletion: Neurofilament hyperphosphorylation
- Clinical
- Onset
- Age: 1 to 3 years
- Nystagmus
- Eye
- Nystagmus: Horizontal & Rotary
- Exotropia (50%)
- Fundi: Pale
- Pupillary reflex: Slow
- Visual acuity: Reduced
- Motor
- Axial hypotonia
- Spasticity: Limbs; Legs > Arms
- Leg muscle wasting
- Seizures (50%)
- Mental retardation: Severe
- Skeletal
- Small head
- Kyphoscoliosis
- Joint contractures
- Progression: Rapid
- Onset
- Brain MRI
- Hypomyelination
- Atrophy: Diffuse
Spastic Ataxia with Optic Atrophy (SPAX4)
● Poly(A) RNA polymerase, mitochondrial (MTPAP; PAPD1)
- Epidemiology: Old Amish family
- Genetics
- Mutation: Homozygous; Missense; N478D
- MTPAP Protein
- Location: Mitochondria
- Function: Polyadenylation of mitochondrial mRNA
- Clinical
- Onset
- Age: Early childhood
- Delayed speech & walking
- Cerebellar
- Ataxia: Limb, Truncal & Gait
- Nystagmus
- Spasticity
- Tongue: Slow movements
- Paraparesis
- Lower limb tone: Increased
- DTRs
- Knee & Jaw: Brisk
- Arm & Ankle: Reduced
- Plantar response: Extensor
- Dysarthria: Cerebellar & Spastic
- Strength: Normal
- Eye: Optic atrophy
- Cognitive: Learning difficulties; Emotional liability; IQ ~50
- Course: Progressive decline in function
- Onset
Spastic Paraparesis with Brain Stem & Spinal Cord Hypomyelination (Leukoencephalopathy) (HBSL)
● Aspartyl-tRNA Synthetase (DARS)
- Epidemiology: 10 patients
- Genetics
- Mutations: Missense; Met256Leu, Ala274Val, Asp367Tyr, Arg460His, Arg487Cys, Arg494Gly, Arg494Cys
- DARS protein
- Expression: High in cortex, Hippocampus + Cerebellar molecular layer & Purkinje neurons
- See
- Clinical
- Onset: 1st year
- Developmental delay: Global; Hypotonia
- Upper motor neuron
- Spasticity: Legs
- Tendon reflexes: Brisk in Arms & Legs
- Plantar: Extensor
- Cerebellum: Nystagmus; Dysarthria (30%); Other usually normal
- Cognition: Mild mental retardation or Normal
- Eye: Optic pallor (30%)
- Course: Progressive; Patients never walked
- Laboratory
- MRI: White matter pathology
- Cerebrum: General atrophy
- Corpus callosum: Increased signal; Thin
- Cerebellar peduncles & Brain stem
- Spinal cord: Corticospinal tracts & Dorsal columns
- NCV: Normal
- MRI: White matter pathology
Progressive Spastic Ataxia & Hypomyelinating Leukodystrophy (SPAX8)
● NK6 Homeobox 2 (NKX6-2)
- Epidemiology: 12 families
- Genetics
- Mutations
- Types: Missense (Arg101Ser; Leu163Val; Phe167Leu; Ala182Val) or Nonsense
- Middle East: 196delC
- Mutations
- NKX6-2 Protein
- Transcriptional repressor with early
- CNS: Early high general & late focused expression
- Regulates oligodendrocyte gene expression
- Clinical
- Onset age: 1 month to 5 years
- Hypotonia: Early
- Cerebellum
- Eyes: Nystagmus; Hypometric saccades; Horizontal EOM Limited; Oculomotor apraxia
- Ataxia: Arms & Legs
- Pyramidal features
- Spasticity
- Tendon reflexes: Increased
- Truncal & Limb
- Legs > Arms
- Dysarthria
- Dystonia (60%): Cervical; Limbs
- Epilepsy
- Polyneuropathy: Distal weakness
- Sensation: Normal
- Cognitive impairment: With early onset disease
- Disease course: Progressive until CNS myelination is complete, then stable
- Laboratory
- Brain MRI
- Hypomyelination: Increased T2W & FLAIR signal in white matter
- Cerebellar atrophy
- Spinal cord: Volume loss; Ventral & dorsal horn T2W hyperintensity
- NCV: Normal motor & sensory
- Brain MRI
5,10-Methylenetetrahydrofolate Reductase Deficiency
● MTHFR
- Genetics
- Polymorphisms C677T & A1298C: Increased sporadic ALS risk in females
- MTHFR protein
- Methyl donor for homocysteine re-methylation to methionine
- Clinical: Varied syndromes
- Common childhood
- Clinical
- Developmental delay
- Psychiatric features
- Polyneuropathy: Some patients
- Encephalopathy
- Nitrous oxide sensitivity
- Laboratory
- Plasma methionine: Low or Normal
- Homocystinuria
- Clinical
- Spastic paraparesis + CNS
80
- Epidemiology: 28 patients; 17 families
- Genetics
- Mutations: Often misense
- Clinical
- Onset age: Adult
- Spinal syndrome
- Spasticity: 4 limbs
- Incontinence
- Slow Progression
- CNS
- Behavior disorders
- Seizures
- Systemic: Pulmonary embolism
- Laboratory
- Fasting homocysteine: High
- Muscle biopsy: Denervation; Complex I deficiency
- Treatment
- Betaine (50 mg/kg twice daily)
- Thiamine (10 mg)
- Riboflavin (100 mg)
- Neural tube defects, Folate sensitive
- Common childhood
Hereditary Sensory Ataxia (SNAX1; ADSA)
● Ring Finger Protein 170 (RNF170)
- Epidemiology
- 4 families
- Genetics
- Mutation: Missense; Arg199Cys
- Allelic disorder: Hereditary Spastic Paraparesis, Recessive
- RNF170 protein
- Expressed in spinal cord
- E3 ubiquitin ligase
- Targets inositol 1,4,5-trisphosphate receptors for degradation
- Other inositol 1,4,5-trisphosphate receptors related SPG syndromes: ITPR1; ERLIN1; ERLIN2
- Clinical
- Onset
- Age: Range 3rd to 8th decade
- Symptoms: Gait disorder; Instability in dark
- Sensory
- Loss: 100%
- Legs > Arms
- Distal
- Pan-modal
- Ataxia (66%)
- Wide based gait
- Romberg: Positive
- Dysesthesias: Some patients
- Loss: 100%
- Reflexes
- Tendon
- Legs: Absent
- Arms: Reduced
- Plantar: Flexor 70%; Extensor 30%
- Tendon
- Motor
- Strength: Usually normal
- Hypotonia in some patients
- Ocular: Abnormal saccades
- Sphincter function: Normal
- Cognition: Normal
- Skin: No ulcerations
- Course: Normal life span
- Onset
- Differential diagnosis
- Laboratory
- Nerve conduction studies
- SNAPs: Normal or Reduced amplitude
- CMAPs: Normal
- H-reflexes: Absent
- SSEP: Legs absent; Arms present
- EMG: Normal
- MRI of Brain & Spine: Normal or Posterior column involvement
- CSF: Normal
- Vestibular areflexia
- Pathology: Spinal cord
- Axonal spheroids in dorsal columns
- Spheroids may contain ubiquitin
- Nerve conduction studies
- Also see: Sensory ataxia syndromes
- RNF170 variant syndrome: Hereditary Spastic Paraparesis 85 (SPG85)
124
- Epidemiology: 6 families
- Genetics
- Inheritance: Recessive
- Mutations: Homozygous; Stop or Missense; Ala109Asnfs*9, Cys102Arg, delEx4_7, Arg173Asnfs*49, Tyr114*, c.396+3A>G
- Allelic disorder: Sensory Ataxia, Dominant
- RNF170 protein
- Mutation effect: Reduced ubiquitination & proteasomal degradation of IP3R
- Clinical
- Onset age: 2 to 5 years
- Spinal cord
- Spasticity: Legs ≥ Arms
- Plantar response: Extensor
- Tendon reflexes: Increased, Legs ≥ Arms
- Urinary bladder function: Usually normal
- Eye: Optic atrophy; Saccadic pursuit
- Ataxia: Some patients
- Sensory: Subclinical involvement of sensory tracts
- Course: Slow progression; Loss of ambulation in 3rd or 4th decade
- Laboratory
- NCV: Axon loss in 2 patients
- Brain MRI: Normal or Cerebellar atrophy
Spasticity, Childhood-onset, with Hyperglycinemia (SPAHGC)
● Glutaredoxin 5 (GLRX5)
- Epidemiology: Lebanese & Chinese families
- Genetics
- Mutations: K51del; 8-BP ins, NT82
- Allelic disorder: Sideroblastic anemia, pyridoxine-refractory 3 (SIDBA3)
- GLRX5 protein
- Mitochondrial protein
- Iron-sulfur (Fe-S) cluster biogenesis
- Maintenance of intracellular iron homeostasis
- Mitochondrial Fe-S cluster transfer to apoproteins (e.g. lipoate synthetase)
- Clinical
- Onset age: 10 months to 2.5 years
- Pyramidal
- Spastic diplegia: Legs > Arms
- Plantar response: Extensor
- Tendon reflexes: Increased
- Ataxia
- Cognitive: Learning difficulties or Normal
- Optic atrophy
- Laboratory
- Brain MRI: Leukodystrophy; C-spine lesions
- Hyperglycinemia
- Respiratory chain enzymes: Normal
Neurodegeneration with Brain Iron Accumulation 7 (NBIA7)
● RALBP1-associated EPS domain-containing protein 1 (REPS1)
- Epidemiology: 1 French family, 2 patients
- Genetics
- Mutations: Compound heterozygous; Missense; V78L, A113E
- REPS1 protein
- Interacts with RALBP1
- Forms endosome recycling compartment
- Interacts with RALBP1
- Clinical
- Onset age: 1 to 10 years
- Motor delay
- Ataxia: Nystagmus, Dysarthria, Dysmetria
- Spasticity: Legs
- Pes cavus
- Course: Progressive
- Laboratory
- Brain imaging: Cerebellar & Cerebral atrophy, Corpus callosum thin, Iron accumulation in globus pallidi & peduncles
Neurodegeneration with Brain Iron Accumulation 8 (NBIA8)
● Carnitine acetyltransferase (CRAT; CAT1)
- Epidemiology: 1 Turkish patient
- Genetics
- Mutation: Missense; Homozygous; Arg321His
- Possible allelic disorder: Carnitine acetyltransferase deficiency
- CRAT protein
- Fatty acid metabolism
- Mitochondrial: Inner membrane
- Catalyzes reversible transfer of acyl groups from acyl-CoA thioester to carnitine
- Control of acyl-CoA/CoA ratio in mitochondria, peroxisomes, & endoplasmic reticulum
- Clinical
- Onset age: 2 years
- Speech delay
- Hypotonia
- Ataxia: Gait disorder; Tremor; Dysmetria
- Spasticity: Tendon reflexes brisk
- Neuropathy: Sensory
- Course: Progressive
- Laboratory
- Brain imaging: Cerebellar atrophy; Basal ganglia hyperintensities consistent with iron accumulation
Neurodegeneration with Brain Iron Accumulation 9 (NBIA9)
● Ferritin heavy chain 1 (FTH1)
- Epidemiology: 5 patients
- Genetics
- Mutations
- Heterozygous
- Truncating
- Duplication or Deletion
- Location: Exon 4 (last exon) of FTH1
- Allelic disorders
- Hemochromatosis 5
- Pontocerebellar Hypoplasia + Neuroferritinopathy 157
- Hemochromatosis 5
- Mutations
- FTH1 protein
- Ferritin subunit
- Ferroxidase activity: Fe++ → Fe+++
- Allows iron storage ferritin mineral core
- Binds to: TFR1
- Mutation effect: Dominant negative
- Clinical
- Onset: Childhood
- Developmental delay
- Spasticity
- Ataxia
- Dystonia
- Startle response
- Microcephaly
- Laboratory
- Serum iron: Borderline low or Normal
- Brain MRI
- White matter: Loss, Corpus callosum thin
- Cerebellar atrophy
- Basal ganglia: Iron
- Pontocerebellar hypoplasia
- Brain pathology: Inclusions, eosinophilic, FTH1+; Iron deposition
Spastic-Ataxia: Neurodevelopmental disorder with poor Growth, absent Speech, progressive Ataxia & Face Dysmorphism (NEDGSAF)
● Suppressor of LIN12-Like (SEL1L)
- Epidemiology: 1 Moroccan family
- Genetics
- Mutatation: Missense, MET528ARG; Homozygous
- Allelic disorder: Neurodevelopmental disorder with hypotonia, poor growth, dysmorphic facies & agammaglobulinemia
- SEL1L protein
- Component of protein complex
- Retrotranslocation or dislocation of misfolded proteins from ER lumen to cytosol
- ERAD-associated
- Clinical
- Onset age: Early infancy
- CNS
- Delay: Global development & motor
- Intellectual development: Severely impaired
- Seizures: Few
- Ataxia: Wide-based & steppage gait
- Spastic paraplegia: Later onset; Legs
- Dystonia: Mild
- Skeletal
- Short stature
- Microcephaly
- Scoliosis
- Pes cavus
- Face: Dysmorphic, Ocular anomalies
- Laboratory
- Brain MRI: Periventricular cavities; Corpus callosum thin
Hereditary Spastic Paraplegia, Complicated (SPG82)
● CTP:phosphoethanolamine cytidylyltransferase (ET; PCYT2)
- Epidemiology: 10 families
- Genetics
- Mutations
- Types: Missense & Stop
- Cys30Gly, His244Tyr, Pro307Leu, Arg377Ter
- General: Most missense in 2nd catyalytic domain; Few in 1st catalytic domain
- Allelic disorder: CNS & PNS Neurodegeneration in Sarlooswolfdogs
- Mutations
- PCYT2 protein
- Etherlipid biosynthesis
- Kennedy pathway: Phosphatidylethanolamine synthesis
- Via CDP-ethanolamine pathway
- Rate limiting enzyme
- Via CDP-ethanolamine pathway
- Strongest expression: Liver, Heart & Skeletal muscle
- Phosphatidylethanolamine: Abundant membrane lipid
- Other etherlipid disorders: Zellweger syndrome; Rhizomelic chondrodysplasia punctata
- Clinical
- Onset age: < 2 years
- Developmental delay, global, with regression
- Spasticity: Para- or Tetraparesis
- Spasticity
- Tendon reflexes: Brisk
- Plantar response: Extensor
- Epilepsy
- Eye: Nystagmus; Optic atrophy
- Polyneuropathy: Some patients
- Sensory loss
- Weakness: Distal
- Laboratory
- Brain MRI: Cerebral & Cerebellar atrophy, progressive
- NCV: Small CMAP & SNAP ampltudes
Leukodystrophy & Cerebellar atrophy (LDCA)
● LSM7 protein (LSM7)
- Epidemiology: 2 patients
- Genetics
- Mutations: Missense; Asp41Asn, Arg69Trp
- Allelic disorder: Fetal death; Homozygous Arg69Trp
- LSM7 protein
- Sm-like protein
- Pre-RNA splicing regulation
- mRNA degradation
- LSM proteins: Assemble in complexes
- Central pore interacts with short uracil-rich stretches of RNA
- LSM7 complexes: LSM1-7 & LSM 2-8
- LSM1-7: Cytoplasmic factor for mRNA decay
- LSM2-8: Nuclear chaperone protein complex for U6 small nuclear RNA (U6 snRNA)
- U6 snRNA: Spliceosome component; LSM2-8 contributes to its nuclear retention
- Clinical: Spastic ataxia
- Onset: Birth to 3 years
- Spastic quadriparesis
- Intellectual disability
- Cerebellar: Ataxia & Nystagmus in 1 patient
- Other: Hearing loss; Dyskinesia
- Laboratory
- Brain MRI: Cerebellum small; Cerebellar white matter hyperintensity
Spastic dystonia syndromes
- DOPA-responsive dystonia
● GTP cyclohydrolase I; Chromosome 14q22.2; Dominant
● Tyrosine hydroxylase; Chromosome 11p15.5; Recessive
- Mitochondrial:
Hereditary spastic dystonia with Leber's optic atrophy
● NADH Dehydrogenase, Subunit 4
- SPG
- Neuroaxonal dystrophy
- Ataxias
- Triosephosphate isomerase (TIM) deficiency
Mass lesions
- von Hippel-Lindau
:
Hemangioblastoma
- Neurofibromatosis
- Meningioma
- Multiple hamartoma
- Lipoma
Other familial spinal syndromes
| Differential diagnosis of hereditary spastic paraplegia | |
| Category | Disorders |
| Structural abnormality | Spinal:
Spondylosis; Atlanto-axial; Canal stenosis Vascular: Arteriovenous malformation Congenital: Arnold-Chiari; Tethered cord Mass: Neoplasm; Granuloma; Syringomyelia |
| Degenerative disorders | Multiple sclerosis; Amyotrophic lateral sclerosis; Spinocerebellar ataxias |
| Leukodystrophies | Adrenomyeloneuropathy; Krabbe; Metachromatic leukodystrophy |
| Metabolic disorders | Vitamin deficiency: B12; E; Abetalipoproteinemia; Mitochondrial disorders |
| Infections | Syphilis; HTLV; HIV (AIDS); Lyme |
| Other | DOPA-responsive dystonia; Hydrocephalus; Toxins |
Return to Spinal Cord Index
Return to Neuromuscular home page
References
1. Neurology 1996;46:1507-1514, Handbook of Clinical Neurology,2013;113:1899-1912
2. Am J Med Genet 1997;69:33-43
3. Am J Human Genetics 1999;64
4. Neurology 1999:53:50-56, Nature Genetics 2007; Online February, Neurogenetics 2007;8:301-305, Brain 2010 Online January
5. Human Genetics 1999;104:443-448
6. Am J Human Genetics 1999;65:757-763, J Neurol Neurosurg Psychiatry 2008 Feb 1, Hum Mutat 2008 Oct 13
7. Am J Human Genetics 2000;66:702-707, 2002;70, Am J Hum Genet 2008 Jun 18
8. Am J Human Genetics 2000;66:728-733, J Clin Invest 2012; Online Jan
9. Hum Mol Genetics 2000;9:637-644, Am J Human Genetics 2001;68:, Eur J Hum Genet 2009;17:187-194
10. Am J Human Genetics 2000;67
11. J Neurol Neurosurg Psychiatry 2000;69:150-160, Ann Neurol 2016 Feb 9
12. Am J Med Genet 2000;94:5-8
13. Am J Med Genet 2000;94:1-4
14. Am J Human Genetics 2000;67
15. Neurology 2001:56:A323&A324
16. Neurology 2001:56:1230-1233, Am J Hum Genet 2008 Online April
17. Brain 2001;124:1426-1437
18. Am J Med Genet 2001;101:135-141, European Journal of Human Genetics 2010; Online June, Pediatr Nephrol 2021;36:3571-3583
19. Nature Genet 2001;29:326-331, Arch Neurol 2004;61:1867-1872, Hum Mol Genet 2022 Aug 4
20. JNNP 2001;71:795-797
21. Am J Human Genetics 2002; March, Am J Human Genetics 2012;91:548¢552
22. Hum Mol Genetics 2002;11:153-163
23. Neurology 2002:58:411-416
24. J Neurol Sci 2002:195:71-76
25. Neurology 2002;58:43-47
26. Ann Neurol 2002;51:681-685
27. J Med Genet 2002;39:387¢390
28. Nature Genet 2002;31:347-348, J Neurol 2004;251:1105-1110
29. Am J Human Genetics 2002;71:518-527
30. Am J Med Genet 2002;111:152-156, Neurogenetics 2008 May 8
31. Neurogenetics 2001;3:91¢97, Neurology 2009 Online April
32. Am J Human Genetics 2002; November
33. Neurology 2002;59:1905¢1909
34. Am J Hum Genet 2003;72:February
35. Am J Hum Genet 2003;72:February
36. Am J Medical Genetics 2003;117A:116¢121
37. Brain 2003;126:783-791
38. Neurology 2003;60:1529¢1532
39. Neurology 2003;61:235-238, AM J Hum Genet 2008; January
40. Am J Hum Genet 2003;73:September
41. Am J Hum Genet 2003;73:September, J Clin Neurosci 2021;94:281-285
42. Am J Hum Genet 2003;73:October
43. Nature Genetics March 2004, Brain 2004: Online August
44. Am J Hum Genet 2003;April 2004, Eur J Med Genet 2019;62:103708
45. Nature Genetics April 2004
46. Am J Hum Genet 2004;75:July
47. Neurology 2004;63:710-712
48. J Med Genet 2004;41:634¢639, Neurology 2006;67;2239-2242, Brain 2010; Online November, J Neurol Neurosurg Psychiatry 2020 Sep 17
49. Am J Med Genet 2005; Online Jan
50. J Med Genet 2005;42:80¢82, Am J Human Genetics 2013; Online June
51. Ann Neurol 2005;57:567¢571, Am J Human Genet 2012; Online November
52. Ann Neurol 2005;57:730¢737, Hum Mol Genet 2015 Sep 18
53. Spine 1999;24:2456-2460
54. Brain 2004;127:973-980, Hum Mutat 2008;29:522-531, Neurology 2008;71:1500¢1505
55. Ann Neurol 2005; Online Dec 22
56. Brain 2006; Online January 24, Genome Research 2011;21:658¢664
57. Neurogenetics. 2006;7:127-129; Am J Hum Genet 2006; Online May, Brain 2008; Online March
58. Am J Hum Genet 2006; Online June
59. Neurosurgery 2006;58:1027-1039
60. Hum Genet 2006 119:611-616, J Bone Metab 2014;21:127-132
61. Am J Hum Genet 2007;80:152-161
62. Hum Genet 2007 Feb 2
63. Neurology 2007;68:1837¢1840
64. Am J Human Genet 2008; Online March, Muscle Nerve 2011;43:19¢25
65. Chin Med J 2008;121:430-434
66. Am J Human Genet 2008; Online May
67. Joint Bone Spine 2008 Apr 28
68. Neurology 2008; On-line May 7, Hum Mutat 2010 Jan 26
69. Neurology 2008; Online Apr 9
70. Mov Disord 2008 Nov 12
71. American Journal of Human Genetics 2008 Online December
72. American Journal of Human Genetics 2008;83:643¢648
73. Brain 2008; Online Dec 2008
74. Neurogenetics 2009; Online May
75. Neurogenetics 2009; Online Dec
76. Neurogenetics 2010; Online July, Am J Human Genet 2013; Online Jan
77. PLoS Biol 2010;8:e1000408, Neurol Genet. 2016;2:e98
78. Am J Human Genet 2010; Online October
79. Am J Human Genet 2010; Online November
80. JNNP 2010; Online December, Neurol Sci 2026;47:178
81. Am J Human Genet 2010; Online December
82. J Med Genet 2011;48:141-144
83. Neurogenetics 2011; Online Aug, Ann Clin Transl Neurol 2023 Sep 27
84. J Neurol Sci 2011;305:67¢70, Neurogenetics 2012 Jan 31
85. Am J Human Genet 2011; On line November
86. Am J Human Genet 2012; On line June
87. Neurology 2012;79:1507¢1514
88. Ann Neurol 2012;72:510¢516
89. Am J Human Genet 2012; Online November
90. Am J Human Genet 2012; Online November
91. Am J Human Genet 2012; Online November
92. Am J Human Genet 2013; Online Jan
93. Human Molecular Genetics 2013; Online April
94. Am J Hum Genet 2013;92:774-780
95. Brain 2013 Dec 19
96. Am J Human Genet 2014; Online Jan
97. Science 2014;343:506-511, Hum Mol Genet 2015;24:6877-68s85, Parkinsonism Relat Disord 2020;77:70-75, J Clin Med 2020;9:1212, Hum Genet 2024 Oct 4
98. Neurology 2014;83: Online July
99. JAMA Neurol 2014;71:470-475, Cerebellum 2023 Jan 25
100. Neurology 2015; Online Feb
101. Int J Neurosci 2015 May 22
102. Brain 2015 May 29, Brain 2015; August
103. J Neurol 2015; Online Sept
104. Neurol Genet 2015;1:e32
105. Eur J Paediatr Neurol 2015 Oct 22, Hum Mutat 2021;42:762-776
106. Brain 2015; Online November
107. J Neurol Sci 2016;362:287-291
108. Cerebellum 2016 Mar 19
109. Am J Human Genet 2016; Online May, Cell Rep 2016 Jun 15, Ann Clin Transl Neurol 2020 Aug 29
110. Brain 2016; Online May
111. Ann Neurol 2016; Online August
112. Brain & Development 2016: Online October
113. Hum Mol Genet 2016;25:2158-2167
114. Brain 2017; Online Jan
115. Am J Human Genet 2017;100:364-370
116. Ann Neurol 2017 Mar 2, J Hum Genet 2024 Feb 14
117. Am J Hum Genet 2017;100:969-977
118. Brain 2018; Online January, Neuron 2018;97:1268-1283
119. Eur J Hum Genet 2018 Feb 16
120. Am J Hum genet 2018; Online March
121. J Neurol 2019 Jan 12
122. Hum Genomics 2019 Apr 16
123. Nat Rev Neurol 2019 Sep 26
124. Nat Commun 2019 Oct 21
125. Brain 2019 Oct 22, Neurol Genet 2022;8:e658
126. Neurol Genet 2019;5(6):e374
127. Am J Physiol Cell Physiol 2020 Apr 29
128. Int J Mol Sci 2020 Nov 2;21(21):8208
129. J Clin Neurol 2021;17:534-540
130. Brain 2020;143:2929-2944
131. J Dairy Sci 2021 Dec 23
132. Neuron 2022 Jan 11
133. J Clin Med 2020;9:913
134. Am J Med Genet A. 2022 Mar 11
135. Cell Rep 2022;39:110598
136. Front Genet 2022;13:936292
137. Ann Clin Transl Neurol 2023 Jan 9
138. Brain Sci 2022;12:1662
139. Brain 2023 Jan 30
140. Ann Clin Transl Neurol 2023 Jul 7
141. J Clin Invest 2023;133:e162836
142. Ann Clin Transl Neurol 2023 Jul 20
143. J Neurosci 2023 Sep 25
144. Front Neurol 2023:14:1301147
145. Brain 2024 Mar 25
146. Mol Genet Genomic Med 202412:e2435
147. J Clin Immunol 2024;45:15
148. Neuromuscul Disord 2025;48:105312
149. Mol Med 2025;31:110
150. Ann Clin Transl Neurol 2025 Apr 25
151. Parkinsonism Relat Disord 2025;137:107946
152. Brain 2025 Aug 14
153. Neurol Sci 2025 Aug 18
154. Am J Case Rep 2020:21:e919463
155. Front Neurosci 2025:19:1684980
156. Nat Commun 2025;16:10537
157. Neurol Genet 2026;12:e200342
158. Nat Commun 2026 Feb 14
2/15/2026