|
Home, Search, Index, Links, Pathology, Molecules, Syndromes, Muscle, NMJ, Nerve, Spinal, Ataxia, Antibody & Biopsy, Patient Info |
Motor Syndromes, Hereditary (SMA, ALS + ...)
|
ALS: Hereditary & Familial Spinal muscular atrophy (SMA): Types Recessive SMA SMA: SMN 5q13 Congenital with arthrogryposis Werdnig-Hoffmann Kugelberg-Welander Spinal muscular atrophy 2 (SMA2) Spinal motor neuropathy: RBM7; 11q232 SMA + Congenital fractures TRIP4; 15q22 ASCC1: 10q22 SMA + Encephalopathy: TBCD; 17q25 SMA + Myoclonus Epilepsy: ASAH1; 8q22 SMA + Pontocerebellar hypoplasia (PCH) PCH1A: VRK1; 14q32 PCH1B: EXOSC3; 9p11 Other: Motor + Cerebellar EXOSC8; 3q13 EXOSC9; 4q27 Mitochondrial DGUOK: 2p13 SCO2: 22q13 SLC25A21: 14q13 TK2: 16q22 Congenital contractures Dominant, Proximal Adult: VAPB; 20q13 Adult + Cramps (SMAJ): CHCHD10; 22q11 Adult: MORC2; 22q12 Bulbar Congenital + Legs weak: TRPV4; 12q24 HMSN-P (Okinawa type): TFG; 3q12 Respiratory & Proximal arms: MAPT; 17q21 Scapuloperoneal syndromes SMALED 1: Leg predominant; DYNC1H1; 14q32 2: Early-onset; Contractures; BICD2; 9q22 X-linked SMA (Recessive) Bulbospinal (Kennedy): AR; Xq12 SMAX 2: Infant + Arthrogryposis; UBE1; Xp11 3: Distal; ATP7A; Xq21 |
HMN D1: UBE3C; 7q36 D2 (2A): HSPB8 (HSP22); 12q24 D3 (2B): HSPB1 (HSP27); 7q11 D4 (2C): HSPB3 (HSPL27); 5q11 D5 (5A): GARS; 7p15 D6 (2D): FBXO38; 5p31 D7 (7A): SLC5A7; 2q12 D8: Congen, Legs: TRPV4; 12q24 D9: WARS1; 14q32 D10: EMELIN-1; 2p23 D11: SPTAN1; 9q34 D12 (5B): REEP1; 2p11 D13 (5C): BSCL2; 11q13 D14 (7B): DCTN1; 2p13 R1 (6): IGHMBP2; 11q13 R2: SIGMAR1: 9p13 R3: 11q13 R4: PLEKHG5; 1p36 R5: DNAJB2 (HSJ1); 2q35 R6: REEP1; 2p11 R7: + Myopathy; VWA1; 1p36 R8: SORD; 15q21 R9: COQ7; 16p12 R10: VRK1; 14q32 + Upper motor neuron Senataxin; 9q34; Dom 4q34: Dom HMN R2: 9p21 D3: HSPB1; 7q11 D13: BSCL2; 11q13; Dom D14: Dynactin; 2p13; Dom SPG + Motor neuropathy HMN: 11p; Rec HMN: 16p HMN: KCC3; 15q14; Dom HMN: MME; 3q25; Rec HMN: SLC5A6; 2p23; Rec HMN: BANF1; 11q13 HMN: MAPT; 17q21 CMT 2N: AARS; 16q22; Dom CMT 2O: DYNC1H1; 14q32; Dom Neuromyotonia: HINT1; 5q31; Rec Child: BICD2; 9q22; Dom Intellect Disability: TBCK; 4q24; Rec |
Motor Neuropathy Differential diagnosis General, Distal Lower motor neuron Motor syndromes Protein mechanisms Also: NMJ disorders Distal SMA (DSMA; dHMN) Recessive dHMN: SYT2; 1q32 + Ataxia telangectasia: ATM; 11q22 + Encephalopathy: TBCE; 1q42 Lethal congenital contractures NRCAM: 7q31 Dominant Calf predominant: FBXO38; 5p31 Leg predominant Distal Ulnar-Median Childhood: BICD2; 9q22 + Macular Δ: FBLN5; 14q32 + Hearing loss: MYH14; 19q13 Scapuloperoneal: TRPV4; 12q24 SMAJ1: GARS1; 7p14 HMN: HARS; 5q31 HMN: GBF1; 10q24 HMN: YARS1; 1p35 X-linked dHMN: AIFM1; Xq26 SMAX 3: ATP7A; Xq21 SMARD2: LAS1L; Xq12 Mitochondrial mtATP6 ± Episodic weakness mtATP8 Sporadic: Hirayama Distal Motor Neuropathy or Myopathy |
Multisystem disorders Recessive AAA syndrome: Aladin; 12q13 ANE: RBM28; 7q31 Chediak-Higashi: LYST; 1q42 COMNB: SLC5A6; 22p23 CONDCA: AGTPBP1; 9q21 CONDSIAS: ADPRHL2; 1p34 Hexosaminidase A: HEXA; 15q23 Leukoencephalopathy: SCP2; 1p32 MND + Dementia & Ophthalmoplegia MPAN: c19orf12; 9q12 NEDCAM: GEMIN5; 5q33 Optic atrophy: c19orf12; 19q12 STAT5B: 17q21 TBX5: 12q24 Dominant Cataracts & Skeletal abnormalities DDPAC: MAPT; 17q21 HMN: EMELIN-1; 2p23 Machado-Joseph: Ataxin-3; 14q32 Myopathy + Paget: HNRNPA2B1; 7p15 X-linked Cabezas: CUL4B; Xq23 Neuroaxonal dystrophy 2 Polyglucosan body: GBE1; 3p12 Mitochondrial: SCO2 Sporadic: Camera-Marugo-Cohen Spastic paraparesis Spastic paraparesis + Motor neuropathy Bulbar syndromes AAA syndrome: Aladin; 12q13; Recessive Brown-Vialetto-van Laere: SLC52A2/A3 BSMA: Androgen Receptor; Xq12 BSMA: Dominant Bulbar ALS Fazio-Londe: Recessive or Dominant FOSMN PLS, Juvenile: Alsin; 2q33; Recessive Worster-Drought |
Distal SMA
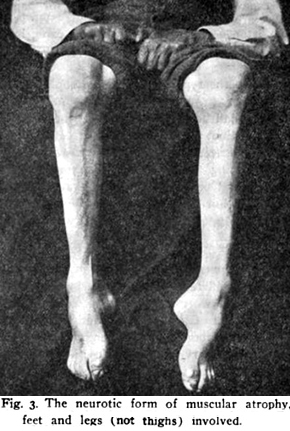 From: Spiller |
HMN: Protein mechanisms
dHMN: Spain 167
- General
- Prevalence: 2.3 per 100,000
- Many sporadic
- Onset age: Often in 1st decade
- Types
Spinal Muscular Atrophy (SMA0; SMA1; SMA2; SMA3; SMA4; SMA 5q)
 178
178
●
Survival Motor Neuron 1 (SMN1)
|
Epidemiology & History Genes Clinical correlations SMN1 SMN2 Modifiers Neighboring & Related SMN Protein Clinical features Congenital Arthrogryoposis SMA 0 Types: 1; 2; 3; 4 Lower motor neuron Pathology Treatments |
|
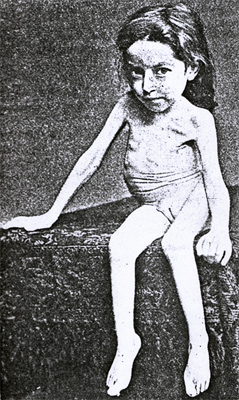 Hoffman ~1891 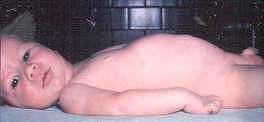 From: Andrew Kornberg MD |
 Guido Werdnig
 Johann Hoffman
|
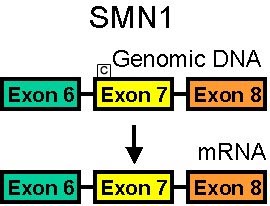
SMA 5q: Classification (without treatment)
- Inheritance: Recessive
- SMN1 mutations: Bi-Allelic
| SMA Type |
SMN2 Copies |
SMA 5q % |
Onset Age |
Motor Milestone Achieved |
Life Expectancy |
| 0 | 1 | < 1% | Birth | Never Sit | < 6 mo |
| 1 | 2-3 | 55% | 0 to 6 mo | Never Sit | 8 to 24 mo |
| 2 | 2-4 | 30% | 6 to 18 mo | Sit | 2 to 4 decades |
| 3 | 3-5 | 10% | 1.5 to 20 yrs | Walk | Normal |
| 4 | 3-5 | 5% | Adult | Walk | Normal |
- History
- SMA described independently by Guido Werdnig & Hoffmann in 1891
- Werdnig
- Described condition as "Neurogenic dystrophy"
- Hoffmann
- Established spinal nature of SMA
- Coined term: "Spinale muskelatrophie"
- Epidemiology
- Incidence of SMA disease: 1 in 6,000 to 20,000 births 25
- 2nd most frequent autosomal recessive disease of childhood (After cystic fibrosis)
- Carrier frequency of SMN1 mutations
- General Western population: 2% to 3%
- Sub-Saharan Africa
- Frequency of carriers lower: 0.05%
- SMN1 copy numbers: Higher; 53% with 3 copies vs 6% with 3 in West
- SMN2 copy numbers: Lower; 24% with none vs 8% in West
- SMN1 (Telomeric SMN (SMNT)) gene
- SMN1 mutations
172
- Most (95% to 98%)
- Deletion of SMN1 gene: Often deletion of entire gene
- Homozygous in 93%
- Few (4%): Deletion of exon 7; May be de novo
- Rare
- SMN1 point mutation (2%): Missense or In-frame deletion common; Also splice site & stop
- Exon 8 deletion
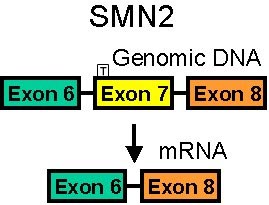
- Gene conversion of SMN1 to SMN2 (Exon 7 C>T)
- Large deletion: SMN1 ± neighboring NAIP gene
- Most (95% to 98%)
- Disease relations
- Number of SMN1 gene copies
8: Varied
- 1 SMN1 copy on each chromosome
- 82% to 96% of normal individuals
- 2 SMN1 copies on one chromosome (Duplication)
- 4% to 18% of normal individuals
- May have deletion on other chromosome
- Variability makes heterozygote testing complicated
- 1 SMN1 copy on each chromosome
- SMN1 gene structure: Strongly homologous to SMN2
- Contains 9 exons
- Exons 7 & 8 contain gene-specific nucleotide sequences
- Some differences from SMN2
- AG-rich exonic splice enhancer in SMN1 exon 7
- Enhancer increases inclusion of exon 7 in protein
- SMN1 gene chromosomal surrounding DNA
- Composition: Large inverted duplication
- Duplication has telomeric (t) & centromeric (c) copies
- Each copy of duplication contains at least 4 genes
- SMN
- Other: p44 (GTF2H2); NAIP; H4F5
- Repeated DNA unit features
- Present on each chromosome
- 0 to 4 copies
- Telomeric (SMN1; SMNT) gene mutations
- Related to presence of SMA disease syndromes
- SMN1 mutations
172
- SMN2 gene (Centromeric SMN (SMNC))
- Gene location: Centromeric to SMN 1 gene
- Strong homology to SMN1: 5 nucleotide differences
- Introns: One difference in 6; Two in 7
- Exons: 2 Differences
- Exon 7 (C to T): Translationally silent
- Exon 8: 3' untranslated region
- Nucleotide differences from SMN1 produce altered SMN splicing
- Can produce identical amino acid sequences to SMN1
- If fully translated
- Quantitative: 10% of SMN1 gene
- Often produces smaller SMN proteins than SMN1
- SMN2 gene transcript often spliced at exon 5 or 7
- 70% to 80% of SMA2 transcripts lack exon 5 and/or exon 7
- Mechanism of altered splicing
- Single-nucleotide change from SMN1 to SMN2 occurs in heptamer motif of the exon splicing enhancer
- New nucleotide sequence (C to T)
- Creates exonic splicing silencer: Inhibition is hnRNP A1
dependent
37
- ? Eliminates motif recognized by splicing factor
- Heptamer motif is recognized by: Splicing factor, arginine/serine-rich (SF2/ASF)
- Nucleotide change abrogates SF2/ASF-dependent exon splice enhancer
- Heptamer motif is recognized by: Splicing factor, arginine/serine-rich (SF2/ASF)
- Creates exonic splicing silencer: Inhibition is hnRNP A1
- Can produce identical amino acid sequences to SMN1
- SMN2: Disease relations
202
- Number of Copies: More copies related to less severity of SMA
- SMN2: Gene number
8: One or Multiple copies
- Normals: No SMN2 copy 9%; 1 SMN2 copy 42%; 2 SMN2 copies 46%; 3 SMN2 copies 3%
- SMA carriers: No SMN2 copy 1%; 1 SMN2 copy 18%; 2 SMN2 copies 47%; 3 SMN2 copies 31%; 4 SMN2 copies 3%
- SMA patients
- 2 or more SMN2 copies
- Reduced SMN2 Gene number correlates with increased SMA severity
- Homozygous SMN2 Deletions: Disease associations
- More common in Lower motor neuron syndromes & MMN
- Sporadic ALS: No clear effect
- SMN2: Disease-relation modifiers
- c.835�549A>G & c.835�44A>G variants: Milder SMA disease
- c.859G>C substitution: SMA less severe
- Plastin 3 (PLS3)
- Ca2+-dependent protein
- Overexpression
- Protective in SMA
- Improves endocytosis
- Neurocalcin Delta (NCALD)
- Protein
- Neuronal calcium sensor
- Interacts with: Clathrin
- Endocytosis suppressor
- Suppression
- Mutations: 17-bp deletion upstream of NCALD; SNP rs147264092 in intron 1
- Clinical effect: Protective
- SMN1 mutations with 3 or 4 copies of SMN2
- Patients continue to be photosensitive
- Epidemiology: 2 families
- Protein
- SMN1 chromosomal locus: General
- Complex 500 kb region of Chromosome 5q13
- Contains large- and small-scale repetitive genetic elements
- Locus of unusual genetic instability
- Frequent spontaneous mutations
- The 500 kb region contains 4 genes: SMN, NAIP, p44, H4F5 (SERF1a)
- The 3 genes are located in a locus of duplicated and inverted DNA (6 genes)
- Telomeric genes are functional genes
- Centromeric genes represent dysfunctional copies
- Genetic structure is unique to humans

- Neuronal Apoptosis Inhibitory Protein (NAIP)
- Females: Absence of NAIP strongly associated with severe phenotype
- Males: No relation between NAIP deletion & phenotype
- GTF2H2 (Btf2-p44)
- Deleted in 15% of SMA patients
- SERF1A (C212/H4F5)
- C212: Multicopy microsatallite marker in an intron of H4F5
- Reduction or absent alleles in
- SMA type I: 94%
- SMA type II: Frequency between type I and controls
- SMA type III: Slightly more frequent than controls
- Closest gene to SMN
- Function unknown
- Retrotransposon-like sequences
- Transcribed into RNA
- Reverse transcribed into cDNA
- Reintegrated as cDNAs into the genome at a new location
- 294 amino acids
- SMN Expression
- From both SMN1 & SMN2 genes
- Transcript splicing
- SMN1 (SMNT) proteins
- 90% full length
- 10% missing exon 5
- AG-rich exonic splice enhancer in SMN exon 7
- Increases inclusion of exon 7 in protein
- SMN2 (SMNC) proteins
- Full length: 20% to 40%
- Missing exon 5, or 7, or both: 60% to 80%
- Most SMN2 spliced to generate short isoform of SMN protein without exon 7
- Less stable: Short half-life
- Poor self-oligomerization
- SMN1 (SMNT) proteins
- Anatomical locations
- High levels: Brain & spinal cord; Kidney; Liver
- Moderate levels: Skeletal & cardiac muscle
- Cell locations: Nuclear & cytoplasmic inclusions (gems)
- Nucleus: 34kDa variant
- Localized to gem bodies & nucleoli
- Near nuclear membrane
- Cytoplasm: 38kDa variant
- Especially in motor neurons
- Sub-cellular locations
- Perikarya; Proximal dendrites; Axons
- Motor neurons: May be associated with cytoskeletal elements & mitochondrial membranes
- Most cells: Not associated with organelles
- Nucleus: 34kDa variant
- Levels in tissue
- Highest during development
- Downregulation of SMN: From early postnatal periods to adulthood
- SMN Interacts with other proteins
- General
- Self oligomerization: Via N- & C-terminus (Exon 7)
- SMN & Gemins 2 to 5 form a multiprotein complex
- Assembles in the cytoplasm
- Translocates to nucleus: Located in gems
- Complex function: Cytoplasmic formation, nucleus import, & regeneration of spliceosomal snRNPs
- SIP1 (Gemin 2)
- Strong interaction: forms heteromeric complex with SMN
- Attachment Via N-terminus of SMN
- Location: Gem bodies in nucleus
- Localization
- Colocalizes with SMN in nucleus & cytoplasm
- SMN does not colocalize with SIP1 in neurites of motor neurons
- SMN & SIP1 form part of protein complex with Sm core proteins & snRNP U1
- Other Gemins
- Gemin 3 (dp103)
: DEAD box putative RNA helicase
- Gemin 4

- Gemin 5
- Gemin 6
- Gemin 3 (dp103)
- Spliceosomal snRNP core proteins (Sm)
- B/B'; D1-3; E; F; G
- Via central tudor domain
- Needed for U snRNP assembly: 5'cap hypermethylation; Import into nucleus
- Spliceosomal snRNA U1 & U5
- Via N-terminus of SMN
- Located in cytoplasm
- HnRNP protein U
& fibrillarin
:
Located in nucleoli
- Bcl-2
:
Coexpression with SMN
- Provide synergistic protection against Bax
or Fas
induced apoptosis
- Provide synergistic protection against Bax
- Nuclear transcription activator E2 of papillomavirus: Via C-terminus
- Profilin 2
- Motoneuron-specific microfilament-associated, actin-binding protein
- Action: Inhibits polymerization of actin
- SMN interaction: Via Pro5-X17-Pro10-X17-Pro5 motif encoded by exons 4, 5 and 6 of SMN gene
- General
- SMN: General functions
51
- Nuclear: Regeneration of active splicing complex
- Part of complex regulating assembly of spliceosomal U snRNPs (RNA-protein complex)
- Essential component of the spliceosome that catalyses pre-mRNA splicing
- Linked to control of protein synthesis & to expression of new protein isoforms
- Axons
- May play role in axonal transport of mRNA
- Promotes neurite outgrowth and/or neuromuscular maturation
23:
Downregulation causes
- Aberrant guidance of axons with excessive axonal branching
- Reduced growth velocity
- Reduced growth cone sizes
- Anomalous calcium-channel clustering in growth cone
- Abnormal presynaptic motor axon nerve terminals
- Presynaptic defects in synaptic vesicles, mitochondria, active zones, neurofilaments & microtubules
- Axonogenesis: May be related to spliced isoform of SMN protein
- Presynaptic motor axon terminals: Postnatal organization & maintenance 84
- Muscle: SMN knockout produces myopathy
- Disease correlations
- SMN mutant proteins: Reduced RNA-binding activity
- Less SMN protein & gem bodies in more severe SMA types
- Mutations: Impaired endocytosis
- Nuclear: Regeneration of active splicing complex
- SMN: Neuromuscular localization
- Nerve
- SMN accumulates in growth-cone-like structures during neuronal differentiation
- Selective deletion of SMN in nerve causes loss of motor neurons
- Muscle
- SMN concentrated at NMJs
- Cytoplasmic SMN high during NMJ formation & reduced with NMJ maturation
- Selective deletion of SMN in muscle produces myopathy 24
- Nerve
- Splicing functions of SMN: Ubiquitous
- Interacts with both RNA-binding proteins and RNA
- RNA-binding element in exon 2a
- Post-transcriptional nuclear RNA metabolism
- During spliceosomal snRNP biogenesis and pre-mRNA splicing
- Related to spliceosomes
- Spliceosomal snRNPs: SMN-related proteins promote transport from cytoplasm to nucleus
- Inhibition leads to
- Reduced spliceosomal snRNP biogenesis
- ? Reduced conversion of pre-mRNA (with introns) to mRNA (no introns)
- SMN Autoantibodies: Clinical associations
199
- Systemic sclerosis + Myositis
- Calcinosis
- Trigeminal neuropathy
- GI involvement, Lower
- Severe phenotype
- Anti-U1RNP + Mixed Connective Tissue Disease (MCTD)
- Skin: Fingertip pitting scars
- Myopathy
- Myocarditis
- Gastrointestinal, lower
- Systemic sclerosis + Myositis
- SMN1 (SMNT; telomeric SMN gene) mutations & disease
- SMN1 mutations present in 95% of SMA patients
- SMN1 & SMN2 genes: Correlation with disease severity
5q CHROMOSOMES
Typical SMN mutations in SMA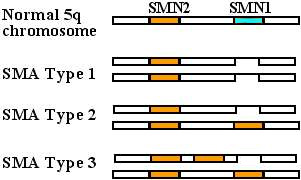
SMN1 Normal gene 
SMN1 Mutation types
Deletion :
More severe SMA
:
More severe SMA
Conversion to SMN2 gene :
Milder SMA
:
Milder SMA
SMN2 gene: Variations
More copies: Correlate with milder SMA.
SMN2 mutations alone: Don't produce SMA- Milder disease (SMA II or III)
- Increased SMN2 gene copy number
- Absence of both SMN1 & SMN2 genes
- Lethal
- SMA type 0: No SMN1 gene & 1 copy of SMN2
- Severe weakness
- Death < 1 month of age
- SMA type I: Mutations
- Mostly SMN1 deletions
- Few missense point mutations in SMN1
- SMN2 gene copy number: Often 2
- SMA type II
- Mutations convert SMN1 gene to SMN2
- SMN2 gene copy number: > 3
- Missense point mutations more common
- SMA type III
- SMN2 gene copy number: > 3
- Missense point mutations more common
- Milder disease (SMA II or III)
- Total amount of full length SMN protein
- ? Best correlation with SMA severity
- SMN1 (SMNT) deletions
- Usually involve exons 3, 6, 7 & 8
- 95% of SMA: Exon 7 of SMN1 gene homozygous absent
- Rare homozygotes for selective 7 & 8 deletion
- Clinically normal, or only mildly weak
- Larger SMN deletions
- Associated with earlier onset & more severe disease
- May affect other neighboring genes
- Hybrids of SMN2 (centromeric)
& SMN1 (telomeric)
SMN genes may occur
- Centromeric exon 7 & telomeric exon 8
- Usually associated with SMA 2 or 3
- Mechanisms producing SMN1 deletions in offspring
25
- Both parents with one SMN1 copy
- SMN1 mutations on one chromosome in each parent
- Recurrence risk for another child with SMA: High (25%)
- De novo rearrangements
- New SMN1 deletion in gene from 1 parent: Parental SMN1 copy number is 2
- Other parent is carrier of SMN1 deletion: SMN1 copy number is 1
- Frequency: 2% of SMA
- Mechanisms of rearrangements: 2 Diferent types
- Unequal crossing over between repeated units during paternal meiosis
- Conversion of part of SMN1 gene into SMN2: Exon 7 of SMN2 & Exon 8 of SMN1
- Recurrence risk for another child with SMA: Low
- SMN duplication in cis: One parent with 2 SMN genes on one allele and 0 on other
- Normal total SMN1 copy number: 2
- 50% chance of transmitting gene with SMN1 deletion
- Recurrence risk for another child with SMA: High (25%)
- Frequency of SMN duplication in cis in SMA parents: 3% to 8%
- Frequency of SMN duplication in cis: 0.1% of population
- Both parents with one SMN1 copy
- Deletions in SMN2 gene alone not related to SMA
- SMN1 (SMNT) missense point mutations
- Frequency: Rare
- 3.6% of 5q13 SMA
- Highest in type III SMA
- Lowest in type I SMA
- ? Higher in some non-European populations: South African blacks
- Other allele: SMN1 deletion
- Location
- Cluster at the 3' end of gene
- Exon 6: Between codons 258 to 279
- Modular oligomerization domain
- Correlation between residual SMN oligomerization (self-association) & severity
- SMN I: Severe loss of function mutations (G279V & Y272C)
- SMN II & III: T274I & S262I
- In tyrosine/glycine-rich motif also present in RNA binding proteins
- Exon 3 termination mutations (425del5; W102X): Exon skipping; Mild phenotype
- Mutation consequences
- Most interfere with SMN oligomerization
- E134K: Alters binding of Sm proteins; Severe SMA phenotype
- Clinical: SMA II & III phenotypes common
- Frequency: Rare
- SMN2 (SMNC)
- Copy number associated with SMA disease severity
- c.859G>C substitution (G287R) 69
- Epidemiology: 3 patients described
- Genetics
- SMA type III patients
- Have fewer SMN2 copies than expected
- Have copies of mutated (G287R) SMN2 gene alone, or in combination with usual SMN2 gene
- Normal population: Mutated SMN2 not common
- SMA type III patients
- SMN protein
- More full length SMN protein produced
- Mutation effect related to splicing modification
- Clinical: SMA less severe than expected from SMN2 copy number
- Proximal strength: Mildly to moderately reduced; Worst in hip flexors & knee extensors
- Patients often SMA type III
- SMN2 Deletions
20: Homozygous
- Genetics
- Mutation: Deletion of exon 7 in SMN2 gene
- Normal population: Homozygous deletion in 5% to 9%
- Increased incidence of homozygous SMN2 deletion in D-LMN syndromes: 36%
- Clinical: Distal lower motor neuron syndromes
- Weakness: Distal; Asymmetric; Upper & Lower extremities
- Course: Rapidly or slowly progressive
- Differences from other LMN syndromes
- Earlier age of onset (40 vs 56 years): Some as young as 15 years
- Lower preponderance of males: M:F ratio of 1.5 vs 2.5
- Both arms & legs involved
- Genetics
- SMN mutations not found in sporadic or familial ALS
- Neuronal Apoptosis Inhibitory Protein (NAIP)
deletions
- Occur in 35% of SMA patients
- In SMA: Virtually always occur with SMN deletion
- Involve exon 5
- Rarely also occur in unaffected
More common in SMA type I (45% - 66%) than in II or III (5% - 16%)- NAIP absence: Correlates with severe phenotype in females, not males
- Rare (2%) SMA 5q patients with neither SMN or NAIP deletion
- Modifier protein: Plastin 3 (PLS3)
- Unaffected SMN1-deleted females: Higher expression
- Onset age: Congenital
- Severe hypotonia
- Movements: Absent; Respiratory failure at birth
- Cranial nerves: Facial diplegia; � External ophthalmoplegia
- Contractures: Especially knees
- Course: Death < 30 days
- Pathology
- Loss of motor & sensory myelinated axons
- Motor neurons: Preserved, swollen
- Rule out X-linked SMA
- Genetics
- Onset age: Prenatal
- Fetal
- Movements: Reduced
- Nuchal translucency
- Gestational age at delivery: Mean 39 weeks
- Muscle weakness: Severe
- Hypotonia: Severe
- Few spontaneous movements
- Respiratory failure
- Cranial nerves: Inability to suck or swallow; Face
- Posture: Frog-like
- Tendon reflexes: Absent
- Tongue fasciculations
- Fetal akinesia deformation sequence
- Contractures: At one or multiple joints
- Micrognathia
- Palate: High arched
- Edema, peripheral
- Intrauterine growth retardation (50%)
- Cardiac
- Congenital defects (85%): Septal
- Bradycardia
- Extraocular movements: Normal
- Course
- Untreated: Death in 1st month
- Treatment
182
- Prolonged survival
- Motor
- Respiratory insufficiency
- GI: G-tube dependent for nutrition
- Weakness: Severe; Some anti-gravity movement
- Skin: Rash; Necrosis & Vasculopathy
- Cardiac: Malformation
- Bones: Fractures
|
|
|
|
|
|
- SMN deletions in > 95%: Exons 7 & 8 tested for deletions
- Carrier testing
- Quantitation of number of SMN1 genes
- Does not detect carriers with > 1 SMN1 gene on a chromosome
|
|
||||||||||||||||||||||||||||||||||||||||||||
Bulbo-Spinal Muscular Atrophy (BSMA; Kennedy's Syndrome; X-linked)
|
Androgen receptor protein Clinical features Clinical-genetic correlations Epidemiology Laboratory features Onset Pathogenic mechanisms Pathology |
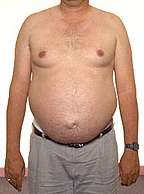
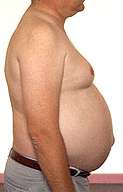 Bulbo-Spinal Muscular Atrophy
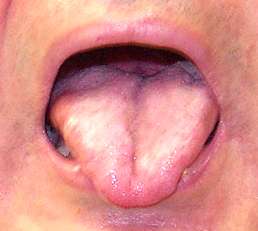
Gynecomastia |
||
|
|
- Epidemiology
- Most common adult onset SMA
- General frequency: 1 in 6,887 to 50,000 192
- SBMA especially common in
- Vasa region of western Finland: Scandanavian founder haplotype
- Some regions in Japan: Founder effect
- Other founder effects
- Different founder haplotypes in various European & Asian countries
- No founder haplotype identified in Canadian patients
- Genetics
- Mutation: Increased CAG repeats
- Allelic disorders
- Androgen insensitivity, Recessive
- Androgen insensitivity, partial ± Breast cancer, Recessive
- Hypospadias 1, X-linked Recessive
- Spinal and bulbar muscular atrophy of Kennedy
- Prostate cancer, susceptibility to, Somatic, Dominant
- Androgen insensitivity, Recessive
- Genetic-Clinical Correlations: CAG repeat length
- Normal CAG repeats
46
- Length: 9 to 39 repeats
- Median lengths in populations
- African American: 19�20
- White Caucasian: 21�22
- Asian: 22�23
- Hispanic: 23
- Location: Exon 1
- BSMA: Long CAG repeats
- BSMA: 40-68 CAG repeats
- CAG repeat length effects
- Longer
- Earlier disease onset
- ? More severe SBMA disease
- Impaired spermatogenesis
- More somatic mosaicism
- No effect on specific clinical features
- Length inversely correlated with transcriptional activity by the androgen receptor
- Longer
- Transmission
- Normal repeat lengths: Transmitted without change; De novo mutations rare
- Expanded repeats: Expansions or contractions in 25% of meiotic events
- Paternal transmission: Larger expansions in CAG repeat number
- Maternal transmission: Contractions or expansions in CAG repeat number
- Prostate cancer
- Fewer CAG repeats (< 18)
- Increased Frequency, especially at young age
- Extraprostatic extension, distant metastases, or high histologic grade
- Prostatic tumor cells
- Dual somatic missense mutations within the AR-CAG repeat
- (CAG)22CAA to (CAG)12CTG(CAG)6CTGCAA
- Interrupts PolyQ tract with 2 leucines
- Dual somatic missense mutations within the AR-CAG repeat
- AR point mutations: Metastatic prostate cancer cells
- Fewer CAG repeats (< 18)
- Androgen insensitivity
- Complete
:
Premature stop codons
- Partial
- Reifenstein Syndrome
:
Point mutations
- Kennedy's syndrome: Bulbo-Spinal Muscular Atrophy
- Reifenstein Syndrome
- Complete
- Other disorders associated with AR-CAG repeat length
- Hereditary hearing impairment
- Schizophrenia
- Benign prostatic hyperplasia
- Risk of developing breast & endometrial cancers
- Normal CAG repeats
46
- Androgen receptor (AR) protein
- Family: Steroid/thyroid hormone receptor
- Phosphoprotein
- Domains
- N-terminal: Contains polyglutamine repeat (Exon 1)
- Central
- DNA binding
- Contains 9 invariant cysteine residues
- Binds zinc ions in 2 zinc fingers
- Nuclear localization signal
- C-terminal
- Binds 2 biologically active ligands
- Testosterone
- Dihydrotestosterone
- Binds 2 biologically active ligands
- Cell locations
- Unbound receptor in aporeceptor complex with heat shock proteins
- Hormone binding
- Receptor translocates to nucleus
- Binds as dimer to DNA through zinc finger domain
- Neuronal function: Plays role in cell survival and dendritic growth
- External link: Androgen Receptor
- BSMA pathogenic mechanisms
- General: Toxic gain of function → Neuronal degeneration
- Large polyglutamine tract: Molecular effect
- Partial proteolysis due to abnormal folding of Androgen receptor
- Truncated forms of androgen receptor may be produced
- Aggregate location: Cytoplasm & Nucleus
- Aggregates
- May contain
- Androgen receptor fragments: Only N-terminal epitopes
- Proteasome components & ubiquitin; PA700 proteasome caps
- Chaperones & Nuclear components with long polyglutamine tracts
- Mitochondria
- Other: Steroid receptor coactivator 1; NEDD8, Hsp70, Hsp90;
HDJ-2/HSDJ; CREB binding protein
- Formation suppressed by HDJ-2 chaperone
- ? Toxic to cell
- Nuclear location of aggregates may be necessary for toxicity
- Truncated forms of AR with expanded repeat
- More toxic than full length protein
- No correlation between frequency of aggregates & cytotoxicity
- May contain
- Proteins possibly involved in mechanism of CAG repeat toxicity
- Caspases: May cleave androgen receptor
- CREB binding protein (CBP)
42
- Polyglutamine-expanded androgen receptor
- Interferes with CBP-mediated transcription of VEGF
- VEGF: May rescue cultured cells with mutant androgen receptor
- Polyglutamine-expanded androgen receptor
- Sir2
pathway
50
- Increased Sir2.1 levels reduce polyglutamine repeat toxicity
- Resveratrol may reduce polyglutamine repeat toxicity
- Loss of Androgen Receptor function
- Non-CAG repeat mutations in androgen receptor reducing function
- Do not produce BSMA by themselves
- Reduced androgen receptor function with CAG expansions
- May exacerbate BSMA disease
- Non-CAG repeat mutations in androgen receptor reducing function
- Clinical features
- Onset
- Age: Mean 27 to 43 years; Range 14 to 75 years
- Early symptoms & signs: Adolescence
32
- Muscle discomfort: Cramps or Pain
- Fatigue: General; Chewing
- Gynecomastia: May be asymmetric
- Weakness: Not common early; May be distal
- Symptoms at 30 years
- Lower > Upper limb weakness
- Occasionally cramps
- Weakness
- Distribution
- Proximal
- Symmetric or Asymmetric (55%; Dominant side 70%)
- Legs > Arms
- Face: Upper & Lower
- Tongue: Weakness; Atrophy; Fasciculations
- Proximal
- Bulbar dysfunction
- Dysphagia: Aspiration
- Dysarthria
- Masseter weakness
- Symmetric
- Cold temperatures: Increased weakness
- Slowly progressive: Over decades
- Distribution
- Other muscle features
- Fasciculation-like movements
- Fasciculations
- Atrophy: Especially face & tongue
- Cramps: 50%
- Tendon reflexes: Absent or reduced
- Sensory: Often subclinical changes
- Vibration: Reduced; Legs > Arms
- Sensory Nerve Action Potentials (SNAPs): Small amplitude
- Tremor
- Hands
- Postural & Action
- Early disease manifestation: 4th decade
- NO upper motor neuron signs
- Systemic
- Nonalcoholic fatty liver disease: Most patients detected by MRS
- Androgen insensitivity related
- Gynecomastia (50% to 70%)
- Reduced fertility: Oligospermia
- Testicular atrophy
- Erectile dysfunction
- Groin hernia: 33%
- Other endocrine
- Diabetes mellitus in some patients: Fasting glucose high in 40%
- Pituitary microadenoma: Rare
- Onset
- Laboratory
- Electrodiagnostic
- NCV
- CMAP amplitude: Reduced in 23% to 40%
- SNAP amplitude: Reduced in 80% in sural
- Distal latencies: Prolonged with cold exposure
- EMG
- Face Twitching: Not typical fasciculations
- Individual discharges: Large amplitude; Asymmetric; Repetitive (Grouped)
- Frequency of discharges: Lower in BSMA (~3/min) than in ALS (~20/min)
- Locations: Face (Especially lower), Tongue, Trunk & Limbs
- Onset: Contraction evoked or Spontaneous
- Electrodiagnostic: Grouped axon discharges (Multiple motor units in each discharge)
- Not: Combined fasciculations, Myokymia or Neuromyotonia
- Fasciculations
- Location: Limbs
- Pattern: Single motor units, Large amplitude potentials
- Neuropathy: Chronic partial denervation with reinnervation
- Fibrillations: Legs > Arms; Less common in arms than in ALS
- Motor units: Large, Long, Polyphasic
- Recruitment: Reduced
- Insertion activity: Increased
- Face Twitching: Not typical fasciculations
- Cortical SSEPs: Absent or Prolonged latencies
- NCV
- Serum
- CK: High (94%); May be elevated early in disease course
- Serum estradiol & gonadotropin: Elevated
- Lipid disorders
- Type II hyperlipoproteinemia
- Type IV hyperlipoproteinemia
- Hypobetalipoproteinemia
- Abnormal
- Total cholesterol (54.7%)
- Low-density lipoproteins cholesterol (40%)
- Triglyceride (48%)
- Muscle biopsy
- Chronic partial denervation
- Grouped atrophy of muscle fibers
- Small angular muscle fibers: Occasional
- Muscle fiber hypertrophy
- NO fiber type grouping
- Internal nuclei
- Type I muscle fiber predominance
- CNS pathology
- Lower motor neurons
- Reduced number in spinal cord & brainstem
- More loss of small motor neurons than ALS
- Sensory ganglion cells: Reduced number in dorsal root ganglia
- Aggregates
- In motor neurons, skin & other tissues expressing androgen receptor
- Intranuclear & Cytoplasmic
- Lower motor neurons
- Electrodiagnostic
- Variant: Patient with 68 CAG repeats
115
- Clinical
- Onset age: 18 years
- Fatigue
- Weakness: Face; Tongue; Proximal; Toes
- Fasciculations
- Cramps
- Tremor
- Dysesthesias: Distal legs & Hands
- Sensory loss: Distal; Panmodal
- Gynecomastia
- Laboratory
- Autonomic: Reduced sweating; Postural tachycardia
- Skin axons: Normal
- Clinical
- Clinical manifestations: Heterozygous females
- General: Mild manifestations later in life
- Muscle cramps: 58%
- Fasciculations: 20%
- Tongue atrophy & fasciculations: 20% in 7th & 8th decade
- Electrophysiology: Chronic denervation in 57%
- Eternal link: e-Medicine
Bulbo-Spinal Muscular Atrophy with Gynecomastia (Autosomal Dominant) 1
● Autosomal Dominant- Onset
- 2nd or 3rd decade
- Nasal voice
- Postural tremor
- Lower motor neuron syndrome
- Tongue: Atrophy; Fasciculations
- Limbs
- Weakness: Mild asymmetry; Proximal + Tibialis anterior
- Atrophy
- Fasciculations
- Tendon reflexes: absent
- Normal: Sensation & Autonomic function
- Cranial nerves: Decreased Upgaze & Convergence (50%)
- Systemic
- Gynecomastia
- No other signs of androgen insensitivity
- Laboratory
- Serum CK: Mildly elevated
- EMG: Acute & Chronic denervation
- Motor evoked potentials (Magnetic stimulation: Prolonged central conduction time
Spinal Motor Neuropathy 150
● RNA-binding motif protein 7 (RBM7)- Epidemiology: 1 Palestinian patient
- Genetics
- Mutation: Homozygous; c.236C>G (p.Pro79Arg)
- RBM7 protein
- Clinical
- Onset
- Age: 1 month
- Hypotonia
- General
- Size (Height, Head circumference & Weight): Low
- Weakness
- Diffuse
- Respiratory
- Muscle: Atrophy; No fasciculations
- Tendon reflexes: Reduced
- Death: 28 months
- Onset
- Laboratory
- Muscle: Grouped fiber atrophy; Large fibers all type 1
- Brain MRI: Normal
Spinal Muscular Atrophy 2 5
● Autosomal Recessive (Not linked to SMA 5q)- Clinical
- Onset: 1 year
- Weakness
- Proximal > Distal
- Symmetric
- Legs > Arms
- Motor functions: Sit but never stand or walk
- Hypotonia
- Muscle atrophy
- Tendon reflexes: Absent
- Tremor: Mild
- Skeletal: Small head circumference; Pes valgus
- Cognitive: Normal
- Progression: ?; May develop respiratory disorders
- Laboratory
- Serum CK: Normal
- Electrophysiology
- EMG: Fibrillations; Large amplitude action potentials
- NCV: Small amplitude CMAPs; Mild slowing; Sensory normal
- Muscle biopsy
- Grouped atrophy
- Type I muscle fiber predominance
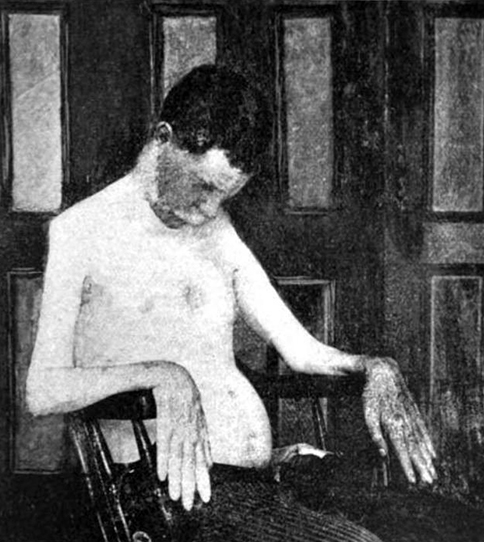
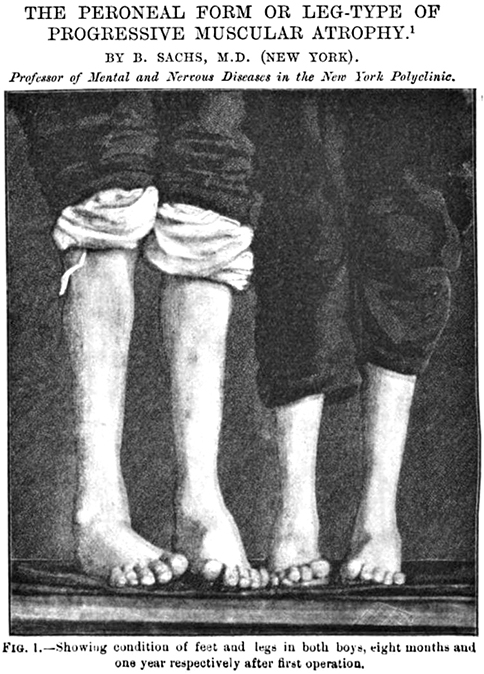
Motor Neuropathies: Hereditary (dHMN & HMN) 49, 85, 152
Hereditary Motor Neuropathies: General features
|
|
Distal hereditary motor neuropathy I (HMND1; Distal HMN I)
59
●
Ubiquitin Protein Ligase E3C (UBE3C)
- Nosology
- Juvenile ALS
- Distal HMN
- Epidemiology: Australian family
- Genetics
- Mutation
- 1.35 Mb complex structural variation
- Intrachromosomal translocation
- Transcript (UBE3C-IF): Lacks 13 of 23 exons of wildtype UBE3C
- Effect: Dominant negative
- Inserted sequence fragment contains
- 4 protein-coding genes & their regulatory elements
- MNX1(HB9)
- NOM1
- RNF32
- LMBR1
- Upstream regulatory elements & first 10 exons of ubiquitin-protein E3 ligase gene E3C
- 4 protein-coding genes & their regulatory elements
- 1.35 Mb complex structural variation
- Allelic disorder: Angelman-like syndrome, Recessive 191
- Mutation
- UBE3C protein
- Ubiquitin E3 protein ligase
- HECT (homologous to E6-AP carboxyl 13 terminus) class of E3 ligases
- Forms thioester complex with ubiquitin in presence of an E1 enzyme & E2 enzyme UBE2D1
- Mutation causes: Duplicated copy of 1st 10 exons
- Clinical
- Onset
- Age: Median 10 years; Range 3�40 years
- Disordered walking & running
- Weakness
- Legs: Ankle extensors & Intrinsic foot muscles
- Hands: Mild or None
- Symmetric
- Atrophy: In regions of weakness
- Upper motor neuron
- Muscle tone: Increased (67%)
- Extensor plantar response (56%)
- Tendon reflexes: Preserved
- Sensory: Normal or Vibration reduced
- Foot deformities: Pes cavus; Hammer toes
- Progression: Slow
- Onset
- Laboratory
- Electrophysiological studies
- Motor NCV: Median normal; Tibial slowed; CMAPs normal
- SNAPs: Reduced with age
- Electrophysiological studies
- Nerve pathology: Axonal loss
- MRI: Normal
Distal hereditary motor neuropathy, type 2 (HMND2; HMN 2A; Distal HMN II)
●
Heat-shock 22-kD protein 8 (HSPB8; HSP22)
- Epidemiology: 4 families
- Genetics
- HSPB8 mutations: Missense; Lys141Asn, Lys141Glu
- Allelic disorders
- HSPB8 protein
- Heat shock protein
- High expression in motor & sensory neurons of spinal cord
- Interacts with HSPB1
- Z-disk-associated CASA complex
- Mutated protein promotes formation of intracellular aggregates
- Clinical
- Onset
- 14 to 35 years
- Weakness of toe extension
- Weakness
- Extensor muscles of feet
- Progression over 5 years to complete paralysis of all distal muscles of legs
- Legs > Arms
- Onset
- Nerve conduction velocity: Normal
- HSPB8 variant syndrome: Motor neuropathy + Distal Myopathy
125
- Epidemiology: 2 families, 7 patients
- Genetics
- Inheritance: Dominant
- Mutations: c.421A>G (p.Lys141Glu); c.151insC
- Allelic disorders
- Clinical
- Onset age: 1st to 3rd decade
- Weakness
- Distal
- Legs > Arms
- Proximal with disease progression
- Cramps
- Fasciculations
- Tendon reflexes: Reduced in legs or Normal
- Sensation: Normal
- Scapular winging: Some patients
- Camptocormia: Some patients
- Course: Slow progression
- Laboratory
- NCV: Motor neuropathy
- Motor axon loss: Length dependant, Legs > Arms
- EMG: Distal leg denervation
- Muscle
- Fiber size: Varied
- Fiber type grouping
- Vacuoles: Rimmed & Non-rimmed; p62 & SMI-31 positive
- Aggregates: Desmin; Myotilin, αB-crystallin; Dystrophin
- Serum CK: 250 to 2,000
- Muscle MRI: Anterior leg involvement
- NCV: Motor neuropathy
- HSPB8 variant syndrome: Limb-Girdle Myopathy with Myofibrillar pathology & Rimmed vacuoles
161
- Epidemiology: 8 families
- Genetics
- Mutations
- Region: C-terminal
- Type: Stop
- c.577_580dupGTCA (p.Thr194Serfs*23); c.525_529del
- Inheritance: de novo; Dominant
- Mutations
- Clinical
- Onset age: 19 to 37 years
- Weakness
- Legs > Arms
- Proximal: Common; Distal in some
- Symmetric
- Respiratory: Some patients
- Paraspinous: Muscle wasting
- Tendon reflexes: Absent
- Sensation: Normal
- Contracture: Posterior neck
- Course: Progressive
- Laboratory
- Serum CK: 600 to 1110
- EMG: Fibrillations; Small motor unit potentials
- Muscle imaging: Paraspinous fatty replacement
- Muscle pathology
- Rimmed vacuoles
- Myofibrillar protein aggregates (Myotilin, Desmin, TIA1)
- Lipid: Increase in type II fibers
Distal hereditary motor neuropathy (HMND3; HMN 2B)
●
Heat-shock 27-kD protein 1 (HSPB1; HSP 27)
- Epidemiology
- Common cause of dHMN
- Families from UK, Croatia, Belgium, Austria
- Genetics
- Missense mutations: R127W; S135F; T151I; P182L
- Mutations in C-terminal domain
- More severe phenotype
- Onset age: 4 to 7 years
- Allelic disorders
- Clinical
- Onset age: 21 to 54 years
- Weakness
- Legs: Distal; Early in course
- Arms: Distal; After 5 to 10 years
- Tendon reflexes: Reduced or Brisk
- Course: Slow progression
- MRI: Anterolateral lower legs relatively spared
Distal hereditary motor neuropathy with upper motor neuron signs 22
● Senataxin (SETX)- Epidemiology: Belgian, Austrian & English families
- Genetics
- Clinical
- Onset age
- 4 to 49 years
- Weakness
- Legs & Arms
- Distal: Intrinsic hands & ankles
- Bulbar: Normal
- Sensory: Usually normal
- Tendon reflexes: Normal or Increased
- Babinski sign: Positive in 50%
- Pes cavus (50%)
- Onset age
- Laboratory
- Electrophysiology
- Nerve conduction velocity: Normal or Mildly reduced
- CMAPs: Reduced amplitude
- Sensory: Normal SNAP amplitude; Conduction velocity borderline or mildly slow
- Central motor conduction latencies: Slow
- EMG: Distal denervation
- Sural nerve pathology: Minor changes
- MRI: White matter changes in 1 patient
- Electrophysiology
- Differential Diagnosis
Distal SMA: Upper limb predominance (HMND5; HMN 5A; SMAD1)
●
Glycyl tRNA Synthetase 1 (GARS1)
- Epidemiology: Multiple ethnic origins
- Genetics
- Allelic disorders
- Mutations: Missense; E71G, L129P, G240R, Leu272Arg, G526R
- Asymptomatic parent: May be mosaic\
- ARS mutations
- GARS protein
- Clinical
- Onset
- Age
- Mean 17 years
- Range: 7 months to 4th decade
- Weakness: Hands
- Age
- Weakness: Varied severity
- Thenar eminence & 1st Dorsal Interosseous
- Lower extremities involved in 50% after 2 years
- Severe cases: Axial; Respiratory
- Tendon reflexes: Reduced
- Cramps: Related to cold exposure or exercise
- Joints: Hyperlaxity
- Rare Pyramidal signs: Rare
- Progression: Very slow to
- Onset
- Laboratory
- NCV: Motor axon loss
- Muscle: SMA-like denervation
Distal SMA: Upper limb predominance (HMND13; HMN 5C)
6
●
BSCL2 gene (Seipin)
- Nosology: Also called HMN 5A
- Epidemiology
- European families
- Most common D-HMN
- Genetics
- Mutations
- Missense
- Exon 3
- Common: Asn88Ser; Ser90Leu
- Located at N-glycosylation site
- Mutation effects
- Alters N-glycosylation site
- Aggregate formation
- BSCL2 allelic disorders
- Mutations
- Seipin protein
- Integral membrane protein: Endoplasmic reticulum
- Glycosylated
- Lipid droplets: Synthesis & ER contacts
- Clinical
- Onset
- Age: Mean 15 to 24 years; Range 2 to 40 years; Childhood often
- Weakness: Hands
- Weakness
- Hands
- Early involvement
- Thenar eminence > 1st Dorsal Interosseous
- Asymmetric
- Lower extremities
- Peroneal weakness (60%)
- Symmetric
- Foot deformities (95%)
- Proximal: Normal
- Hands
- Upper motor neuron
- Tendon reflexes: Brisk
- Tone: May be increased in legs
- Plantar responses: Flexor
- Sensory loss: Vibration reduced in legs
- Hyperhidrosis (40%): Hands & Feet
- Progression
- Very slow: Over decades
- No patients severely handicapped
- Onset
- Laboratory
- Electrophysiology
- CMAPs: Small
- Motor NCV: Normal or Mildly reduced
- Sensory NCV amplitude: Mild reduction, especially older patients
- EMG: Large MUPs; Reduced Recruitment
- Central motor conduction times: Prolonged (60%)
- Sural nerve: Mild loss of myelinated axons
- Muscle MRI: Thenar eminence, Soleus & Tibialis anterior most involved
- Electrophysiology
- BSCL2 variant syndromes
- CMT2-like
99
- Genetics
- BSCL2 Mutation: S90W
- Inheritance: Dominant
- Clinical
- Onset ages: 5 to 30 years
- Weakness: Legs early; Thenar; Distal
- Sensory loss: Pan-modal; Distal
- Spastic: Gait; Tendon reflexes increased (Legs); Extensor plantar response
- Skeletal: Pes cavus
- NCV
- CMAPs: Small amplitudes, especially median
- SNAPs: Small amplitude in some
- Velocities: Normal
- Sensory myelinated axons: Loss or Increased; Regeneration; Thin myelin
- Genetics
- Also see
- CMT2-like
99
Hereditary Distal Ulnar-Median Muscular Atrophy 7
● Autosomal Dominant- ? HMN 5 variant
- Weakness
- Distal
- Arms at onset; Legs later
- Symmetric
- Onset: Childhood - Teens
- Upper motor neuron signs
- Brisk tendon reflexes
- Plantars reflexes: Flexor, Equivocal or Extensor
- Sensory: Normal
- Electrodiagnostic
- Normal nerve conduction velocities
- Prolonged distal latencies
Distal Hereditary Motor Neuronopathy 7A (HMND7; HMN 7A; dHMN-VII; dHMN7) (Vocal cord involvement)
●
Solute carrier family 5 (Choline transporter), Member 7 (SLC5A7; CHT)
- Epidemiology: 4 families
- Genetics
98
- Mutations
- Types: Frameshift or Tuuncating
- c.1497delG (p.Lys499Asnfs*13), p.His521Gln*fs2, p.Lys510Asnfs*2, c.1526del (p.Pro509Leufs*3)
- Allelic disorders
- Congenital MG with Episodic Apnea: Recessive, Missense mutations
- Lethal Congenital Arthrogryposis
- Mutations
- SLC5A7 protein
- Choline transporter
- Presynaptic: Motor neurons
- Determinant of synaptic acetylcholine synthesis & release at NMJs
- Mutation: Reduced choline transport; Remove endoytic trafficking motif
- Clinical
- Onset
- Age: Early childhood to Teens
- Voice or Gait disorder
- Distal weakness
- Onset: Hands; Median distributaion
- Progression to distal leg weakness
- Wasting: Prominent distally
- Usually symmetric but occcasional asymmetry
- Vocal cord involvement (70%)
- Distribution: Often asymmetric; Eventually bilateral
- Onset: 1st or 2nd decade
- Voice: Hoarse; Quiet
- Respiratory failure: 2° Bilateral vocal cord paralysis
- ± Sensorineural hearing loss
- Tendon reflexes
- May be brisk
- Reduced distally in arms & legs with disease progression
- Sensation: Normal
- Course: Slow progression
- Onset
- Electrodiagnostic
- EMG: Distal denervation in feet & hands
- NCV
- Velocities: Normal
- CMAPs: Small distally
- Repetitive nerve stimulation: No decrement
- SFEMG: Excess jitter
- SLC5A7 variant disorder: Congenital MG 20 (CMS20), Presynaptic, with Episodic Apnea
133
- Epidemiology: 6 families
- Genetics
- Inheritance: Recessive
- Mutations: Missense; Loss of function
- Allelic disorders
- Clinical
- Onset
- Age: Congenital to 2 months
- Apnea
- Weakness
- Bulbar: Dysphonia; Dysphagia
- Face
- Eye: Ptosis; Ophthalmoparesis
- Limbs: Proximal > Distal
- Axial: Neck & Other
- Variability
- Apnea: Episodic
- Fatigability
- Arthrogryposis
- Fingers & Knees
- In more severe syndromes
- Associated with: Hypotonia
- Intellectual disability
- Course: Fluctuating weakness; Improvement with treatment
- Treatment: AChE inhibitors
- Onset
- Laboratory
- Repetitive nerve stimulation decrement: 0% to 70%
- Neuromuscular junctions
- Young patient: Immature, Reduced definition of AChR patches; Polyinnervated
- Older patient: NMJs often Denervated or Remodeled; Butyrylcholinesterase (BChE) increased
Distal Hereditary Motor Neuronopathy (Vocal cord involvement) (HMND14; HMN 7B)
34
●
Dynactin 1 (DCTN1)
- Epidemiology: Single family
- Genetics
- Mutation: Missense; G59S
- DCTN1 allelic disorders
- Perry disease
:
Parkinsonism, Depression, Weight loss, Hypoventilation, TDP-43 immunostaining of brain
- May be associated with
- Progressive supranuclear palsy (K56R)
- Perry disease
- Largest subunit of 10 million dalton dynactin complex
- Binds to
- Microtubules
- Dynein: Cytoplasmic
- Also see: Rab proteins, CMT 2B
- Functions
- Associated with axonal transport of vesicles & organelles
- Blockade of dynactin binding to dynein: Blocks vesicle motility along microtubules
- May promote synapse stability at neuromuscular junctions
- Mutation
- Located in p150Glued subunit
- May distort folding of microtubule binding domain: Reduced tubulin binding
- Induces dynactin self-aggregation & with dynein (in vitro)
56
- Associated with mitochondria
- Aggregation reversed by overexpression of Hsp70
- Onset
- 3 years to Early adulthood
- Respiratory difficulty due to vocal cord paralysis
- Weakness
- Face: Progressive
- Vocal cord paralysis
- Limbs: Distal; Hands then Feet
- Sensory: Normal
- Ankle contracture: With early onset
- Tendon reflexes: May be brisk
- Cognition: Normal
- NCV: Velocity normal, CMAPs small
- EMG: MUPs large & long
- Genetics
- Inheritance: Dominant or Sporadic
- Mutations: Missense; Thr1249Ile, Met571Thr, Arg785Trp, Arg1101Lys; R1275C
- Incomplete penetrance
- Clinical
- Onset
- Age: 46 to 64 years
- Weakness: Arms, Legs, Posterior neck or Bulbar
- Weakness
- Arms, Legs or Bulbar
- May be asymmetric
- Progression: Slow; symptom duration = 4 to > 9 years
- Upper motor neuron signs: Present
- Tendon reflexes: Brisk
- Fronto-Temporal Dementia
- Some patients without motor neuron disease
- 1 family
- Onset
- Laboratory
- EMG: Widespread denervation
- MRI: Normal
Distal SMA: Calf predominant (HMND6; HMN2D)
106
●
F-box only protein 38 (FBXO38; MOKA)
- Nosology: Neuronopathy, distal hereditary motor, type IID
- Epidemiology: 2 families
- Genetics
- Mutation: Cys206Arg
- Allelic disorder: dHMN, Recessive
- FBXO38 protein
- Coactivator of transcription factor Kruppel-like factor 7 (KLF7)
- Regulates genes required for neuronal axon outgrowth & repair
- Coactivator of transcription factor Kruppel-like factor 7 (KLF7)
- Clinical
- Onset
- Age: 13 to 48 years
- Difficulty standing or running
- Weakness
- Initial: Calves
- Distal predominant: Hands, Intrinsic; Feet
- Other weak muscles: Triceps
- Proximal: With disease progression in some patients
- Tendon reflexes: Ankle absent
- Fasciculations
- Course
- Slowly progressive
- Some patients lose ambulation
- Onset
- Laboratory
- NCV
- Motor amplitudes: Reduced
- Sensory: Normal
- EMG: Neurogenic; Fibrillations
- Muscle biopsy: Grouped atrophy; Large fibers types II > I
- NCV
- FBXO38 variant: Distal hereditary motor neuronopathy type (dHMN) IID, Recessive
156
- Epidemiology: Turkish patient
- Genetics
- Inheritance: Recessive
- Mutation: Homozygous; p.Arg526Gln
- Clinical
- Onset: Childhood
- Weakness: Distal; Legs > Arms
- Tendon reflexes: Normal
- Sensory: Normal
- Skeletal: Pes cavus
- Hearing loss
- Systemic: Duplex collective system, Arcuate uterus, Choanal atresia
- Laboratory
- EMG: Denervation, distal
- NCV: CMAP amplitudes small
- Brain MRI: Normal
Distal SMA: Leg predominant 19
● Dominant- Epidemiology: Single Italian family
- Onset: 8 to 30 years; Difficulty with heel walking
- Clinical
- Weakness
- Legs: Distal; Tibio-Peroneal
- Arms: Mild; Later in disease course
- Proximal: Mild; Arms & Legs; Late in course
- Hearing: Sensorineural loss in older patients
- Weakness
- Laboratory
- EMG: Denervation in distal muscles
- Nerve conduction: CMAPS small; Normal NCV
- Muscle biopsy: Chronic denervation
- Auditory evoked potentials: Cochlear hearing loss
- See: Congenital SMA of lower limbs
Distal SMA 3 (HMNR3; DSMA 3)
21
●
Chromosome 11q13.3; Recessive
- Nosology: HMN types III & IV
- Epidemiology
- Lebanese & European families
- Most patients probably from single ancestor
- Genetics: Normal IGHMBP2 gene
- Clinical
- Onset
- Age: Infancy to Early adult
- Distal weakness
- Weakness & Atrophy
- Distal
- Feet > Hands
- Diaphragm: With childhood onset
- Proximal & Trunk: With disease progression
- Cranial nerves: Normal
- Sensation: Normal
- Tendon reflexes: Reduced or Normal
- No upper motor neuron involvement
- Course: Slow progression
- Onset
- Electrophysiology
- EMG: Denervation
- NCV: Normal
- Muscle biopsy: Denervation
Distal HMN: Childhood onset
●
Autosomal Recessive
- Clinical features
- Onset: Early childhood
- Weakness: Distal; ± Quadriceps
- Progression: Very slow; Survival until at least middle life
Distal infantile spinal muscular atrophy with diaphragm paralysis (HMNR1; DSMA1; SMARD1; HMN 6)
●
Immunoglobulin μ-binding protein 2 (IGHMBP2)
- Epidemiology
- > 50 patients
- Up to 1% of early onset SMA
- IGHMBP2 genetics
- Mutations: Missense; Nonsense; Frameshift deletion (exon 5) & Splice donor-site
- Allelic disorders
- DSMA1 (SMARD1; HMN6)
- SMARD, milder
- AR-CMT2S
- IGHMBP2 protein
66
- Transcription factor: DNA binding protein
- ATP-dependent 5' --> 3' helicase: Separates double-stranded RNA & DNA
- Regulates: DNA replication, Pre-mRNA splicing, Transcription ± Translation
- Localization
- Similar to SMN1 protein
- Co-localization
- RNA-processing machinery in both cytoplasm and nucleus
- Ribosomes
- Tissue distribution: Widespread
- Mutations: Impair ATPase & Helicase activity
- Clinical features
- Onset
- Age: Congenital to 2 years
- Intrauterine growth retardation
- Respiratory failure
- Hypotonia
- Respiratory distress: Severe
- Diaphragmatic paralysis
- Intercostal muscles: Relatively spared
- Weakness: Predominantly upper limbs & distal muscles
- Tendon reflexes: Reduced
- Contractures: Occasional; Mild; At knee and ankle
- Fingers: Fat
- Course
- Common: Death or respiratory failure at < 3 months
- Onset
- Serum CK: Normal
- Chest x-ray: Eventration of diaphragm
- Spinal cord pathology
- Upper more severely affected than the lower
- Small anterior roots
- Remaining motor neurons show chromatolysis
- Nerve (Sural): Myelinated axon loss
- Muscle
- Neurogenic atrophy without reinnervation
- Large fibers: Type I
- Genetics
- May have same mutation & family as severe disease
- Clinical
- Onset: 2nd year
- Weakness
- Distal then Proximal
- Dysphagia
- Respiratory
- Sleep hypoventilation
- Vital capacity: Normal
- Survival through childhood
- Epidemiology
- 12% of AR-CMT2
- Asian, European, US
- IGHMBP2 Genetics
- Inheritance: Recessive
- Mutations: Heterozygous; Missense, Stop or Splice; Common p.Cys46*
- Clinical
- Laboratory
- NCV: Axon loss, Motor & Sensory
- Nerve biopsy: Loss of large axons
- Splice donor mutation
- Functional IGHMBP2 expression reduced to 20�25% of controls
- Life spans: 12 to 138 days
- Weakness & Muscle wasting
- Progressive & Severe
- Hind limbs then fore limbs
- No phrenic nerve or diaphragm involvement
- Loss of motor neuron innervation
- Genetic disease modifier: On mouse chromosome 13
- Other tissues involved if IGHMBP2 expression replaced in nerve
- Dilated cardiomyopathy: High serum CK & CK-MB
- Skeletal muscle: Myopathy, mild
Distal Hereditary Motor Neuropathy, Jerash type (DSMA 2; HMNJ)
●
Chromosome 9p21.1-p12; Recessive
- Epidemiology: 1 family
- Genetics
- Locus contains: SIGMAR1 gene
- Clinical
- Onset age: 6 to 10 years
- Weakness
- Distal
- Legs, then Arms within 2 years
- Muscle wasting: Hands & Feet
- Steppage gait
- Tendon reflexes
- Brisk at Knees; 6 to 25 years
- Ankle: Absent
- Upgoing toes: Younger than 15 years
- Laboratory
- Electrophysiology
- Motor: Small CMAPs; Normal NCV
- Sensory: Normal SNAPs
- EMG: Chronic denervation
- Serum CK: Normal
- Biopsy: Sural Nerve
- Mild reduction in number of myelinated axons
- Occasional regeneration
- Biopsy: Muscle
- Grouped atrophy
- Target fibers
- Type I predominance
- Electrophysiology
Distal SMA, X-linked 3 (SMAX3)
41
●
ATPase, Cu++-transporting, alpha polypeptide (ATP7A)
- Epidemiology: Brazilian, Australian & North American families
- Genetics
72
- Mutations
- Missense
- Locations: P1386S; T994I
- Allelic disorders
- Menkes disease
: Loss of function mutations
- Occipital horn syndrome
- Defects that allow residual copper transport
- Often via leaky splice-junction mutations involving noncanonical bases
- Menkes disease
- Mutations
- ATP7A protein
- Copper-transporting P-type ATPase
- Normal copper: Localizes to trans Golgi network in basal copper concentrations
- Elevated copper: Relocates to small vesicles and plasma membrane
- Mutations
- ATP7A protein levels: Normal
- Functions: Impaired ATP7A trafficking in response to copper loading
- Clinical
- Onset
- Age: 1 to 61 years; Older in Australian family
- Foot deformity
- Gait disorder
- Weakness & Atrophy
- Legs
- Early in disease course
- Distal
- Tibioperoneal
- Hands
- Usual: With disease progression
- Early in disease course: 1 patient
- Proximal: Normal
- Temperature sensitive: Occasional patients weaker in cold
- Progression
- Slow
- Ambulation: Remains independent
- Legs
- Tendon reflexes: Variable; Absent diffusely or in legs; May be normal
- Plantar response: Neutral
- Skeletal: Pes cavus
- Autonomic: Normal
- Hair: Normal
- Joints & Skin: Normal
- Heterozygous females: Normal
- Onset
- Laboratory
- Serum CK: Normal
- Electrophysiology
- CMAP amplitudes: Reduced or Absent
- NCV: Mildly reduced or normal
- EMG: Chronic denervation
- SNAPs: Normal
- Pathology
- Nerve (Sural): Normal
- Muscle: Denervation
- Normal
- Catecholamine ratios in plasma
- β-2-microglobulin in urine
Spinal muscular atrophy with respiratory failure (SMARD) 2, X-linked (SMAX) 111
● LAS1-like Ribosomal biogenesis factor (LAS1L)- Epidemiology: 2 patients
- Genetics
- Mutations: S477N, Splice (c. 846 G>C)
- Allelic disorder: Wilson-Turner syndrome
- LAS1L protein
- Function: Ribosomal biogenesis
- Coordinates processing of 45S rRNA at both ends of 2nd internal transcribed spacer
- Clinical
- Onset age: Neonatal
- Weakness
- Distal
- Respiratory failure: Early onset; Diaphragm paralysis
- Feeding difficulty
- Fasciculations: Tongue
- Hypotonia
- Contractures: Mild; Toes & Fingers
- Tendon reflexes: Present
- SMARD: Differential diagnosis
- Laboratory
- Electrodiagnostic
- CMAP amplitudes: Reduced in legs more than arms
- SNAPs: Normal
- EMG: Reduced recruitment, especially distal
- Brain MRI: Normal
- EEG: Normal
- CSF: Normal
- Electrodiagnostic
Distal hereditary motor neuropathy 45
● Chromosome 11p; Recessive- Epidemiology: Southern Italian family
Distal Hereditary Motor Neuropathy (HMND4; HMN 2C)
71
●
Heat-shock 27-kd protein 3 (HSPB3; HSPL27)
- Epidemiology: 4 families
- Genetics
- Mutations: Missense; Arg7Ser; R116P; Y118H
- Allelic disorder: Shoulder girdle weakness, p.L34Ffs*50 (A33AfsX50)
- Other: Small heat-shock protein disorders
- HSPB8: HMN2A & CMT 2L
- HSPB1: HMN2B & CMT 2F
- αB-crystallin: Myofibrillar myopathy & Posterior polar cataracts
- HSF1 antibodies
- HSPB3 protein
- Clinical
- Onset
- Age: 3rd decade
- Weakness: Legs
- Weakness
- Distal
- Legs: Severe at ankles
- Arms: Hands with wasting
- Shoulder-girdle: Stop mutation, heterozygous
- Sensation: Normal or Mildly reduced
- Tendon reflexes: Reduced at ankles
- Onset
- Laboratory
- EMG: Denervation in distal legs & hands
- NCV: Normal velocities; Axon loss
Distal Hereditary Motor Neuropathy with Pyramidal features
● Chromosome 4q34.3-q35.2; Dominant- Epidemiology: Italian family
- Clinical
- Onset
- Age: 25 to 40 years
- Gait Disorder: Spastic
- Weakness & Atrophy
- Distal
- Legs
- Upper motor neuron
- Legs
- Spasticity
- Tendon reflexes: Brisk at knees; Reduced at ankles
- Sensation: Slight vibratory loss in feet
- Cognition & Bulbar: Normal
- Progression: Slow; Walking preserved
- Onset
- Laboratory
- Motor nerve conductions: Velocity normal; Amplitude reduced
- SNAPs: Normal
Spinal Muscular Atrophy: Other
Infantile Spinal Muscular Atrophy with Arthrogryposis (XL-SMA; SMAX2; AMCX1)
● Ubiquitin-activating enzyme 1 (UBE1; UBA1)
- Epidemiology: 8 families
- Genetics
- Mutations
- General location
- Exon 15
- Domain function: Interactions with gigaxonin
- Missense: Met539Ile (c.1617G>T; c.1617G>A); Ser547Gly; Glu557Val
- Synonymous (c.1731 C/T, Asn577Asn): Leads to
- Reduced UBE1 expression
- Altered methylation pattern of exon 15
- General location
- Allelic disorder (Somatic mutations)
- VEXAS
(Autoinflammmation): Met41Val; Met41Thr; Met41Leu
- VEXAS
- Mutations
- UBE1 (UBA1) protein
168
- Expression: All tissues; High levels in motor neurons
- Localization
- Nuclear in normal motor neurons
- Cytoplasmic SMN1-linked SMA motor neurons
- Ubiquitin-proteasome system
- Ubiquitin activating E1 enzyme
- Initiates activation & conjugation of ubiquitin-like
proteins
- Binds to gigaxonin
- Modification of proteins with ubiquitin or ubiquitin-like proteins
- Controls many signaling networks
- Requires
- Ubiquitin activating enzyme (E1)
- Ubiquitin conjugating enzyme (E2)
- Ubiquitin protein ligase (E3)
- Ubiquitin activating enzyme (E1)
- Required for Atg7- & Atg3-independent autophagy
- Clinical features
- Onset: Congenital or Infant
- Weakness
- Early: Hypotonia
- Proximal > Distal: Similar to Werdnig-Hoffmann
- Face: Myopathic faces
- Bulbar
- Speech: Nasal
- Tongue: Fasciculations
- Cramps: Some patients
- Tendon reflexes: Reduced
- Skeletal
- Contractures: Proximal & Fingers; Congenital
- Fractures: Congenital
- Face: Dysmorphism
- Genital: Undescended testes
- Course
- Severe disorders: Usually death < 2 years
- Associated with respiratory insufficiency
- Milder patients: Long lifespan
- Family history: Miscarriages/spontaneous abortions
- Laboratory
- Pathology
- Muscle: Denervation
- Anterior horn cell loss
- Brain MRI: Normal
- Serum CK: Mildly high
- NCV: CMAP amplitudes reduced; Velocities normal
- EMG: Fibrillations; Positive sharp waves
- Pathology
- Female carriers: No symptoms
- Rule out: Congenital 5q-linked SMA
Proximal SMA with Dominant inheritance: Adult Onset (Finkel, Late adult type; SMAFK)
● VAPB
- Epidemiology: Common in Brazil; 200 patients
- Genetics
- Brazil & Portugal mutation: P56S
- Allelic disorder: ALS 8
- Clinical
- Onset
- Age: 30 to 60 years; Mean = 49 years
- Legs
- Weakness & Atrophy
- Proximal
- Respiratory: Some patients
- Gait: Waddling
- Fasciculations
- Tendon reflexes: Reduced (80%)
- Sensory & Bulbar: Normal
- Course: slow progression
- Onset
- Laboratory
- Serum CK high
- EMG: Neurogenic; Positive sharp waves, MUPs long duration & large amplitude
- NCV: Velocities normal; CMAP amplitudes reduced; SNAPs normal
- Muscle biopsy: Neurogenic
Hereditary motor neuropathy, distal 8 (HMND8; HMN8): Spinal muscular atrophy, Congenital, non-progressive, of lower limbs (CSMAA)
● TRPV4
- Epidemiology: Multiple families
- Genetics
- Mutations: Missense; Ser94Leu, Arg269Cys (Also with CMT 2C & SPSMA)
- Allelic disorders
- Clinical
- Onset age: Congenital
- Weakness
- Developmental delay: Motor
- Legs only: Proximal & Distal
- Non-progressive
- Vocal cord paralysis: Some patients
- Tendon reflexes: Reduced in legs
- Contractures
- Arthrogryposis
- Knees & Ankles
- Laboratory
- Serum CK: Mildly elevated
- Electrophysiology
- EMG: Chronic (Giant motor units) & Active denervation
- NCV: Normal motor & sensory
- Differential diagnosis
- Distal SMA of lower limbs
- SMALED: DYNC1H1
- SMALED2: BICD2
- SMA Congenital, non-progressive, of lower limbs: TRPV4
Hereditary Motor Neuropathy, Distal 9 (HMND9; HMN9; dHMN type I)
● Tryptophanyl-tRNA synthetase 1 (WARS1)
- Epidemiology: 5 families; 14 patients
- Genetics
- Mutation: c.770A>G (p.His257Arg) (Recurrent); Phe138Tyr (de novo); Asp314Gly (Older onset)
- Allelic disorder: Microcephaly + Developmental delay & Brain anomalies, Recessive 187
- WARS1 protein
- Function
- Cytoplasmic Amino-acyl tRNA synthetase (ARS)
- Catalyzes aminoacylation of tRNAtrp with tryptophan
- Mutation effect: Dominant negative
- Function
- Clinical
- Onset
- Age: 9 to 23 years
- Weakness: Distal; Legs
- Gait disorder
- Weakness & Wasting
- Distal
- Arms & Legs
- Moderate to Severe
- Sensation: Normal
- Tendon reflexes: Ankles absent; Knees absent or normal
- Feet: High arches
- Course
- Progressive
- Most remain ambulant
- 1 patient in wheelchair
- Onset
- Laboratory
- Electrodiagnostic
- NCV: 35 to 63 M/s
- CMAPs: Reduced amplitude
- SNAPs: Normal amplitude
- Pathology
- Sural nerve: Normal
- Muscle: "Neurogenic atrophy"
- Muscle MRI: Distal atrophy
- Electrodiagnostic
Early-onset spinal muscular atrophy with Contractures (SMALED2A)
● Bicaudal D, drosophila, homolog of, 2 (BICD2)
- Epidemiology: 23 families
- Genetics
- Mutations
- Hot spot: Ser107Leu
- Other: Asn188Thr; Ile189Phe; Val485Gly; Arg501Pro; Lys508Thr;
Ala535Val; Tyr557His; Ser681Leu; Thr703Met; Arg747Cys; Glu774Gly - Locations: Coiled-coil domains
- Allelic disorders
- SMALED2A
- SPG
- Arthrogryposis (SMALED2B)
- Distal myopathy
- Mutations
- BICD2 protein
- Golgin
- Adaptor protein
- Maintains Golgi structure
- Interacts with the dynein-dynactin motor complex: Axon transport
- Facilitates trafficking of cellular cargos involved in motor neuron development & maintenance
- Mutations
- Increased microtubule stability
- Abnormal collateral axon branching
- Impaired NMJ development
- Dynein-related disorders, Other: SMA-LED; CMT 2O
- Clinical
- Onset
- Age: In utero, Congenital to Adult
- Fetal movements reduced
- Walking: Late, Difficult or Slow
- Contractures: Ankles
- Weakness
- Legs > Arms
- Legs: Proximal & Distal
- Arms: Mild; Proximal & Hands
- Respiratory: Sleep disordered breathing in some patients
- Symmetric
- Most patients ambulant
- Muscle wasting: Legs
- Skeletal
- Feet: Pes planus or High arch; Calcaneovalgus
- Hip dislocation: Congenital or Early-onset
- Spine
- Scoliosis (50%): Not severe
- Lordosis
- Contractures
- Common: Not all patients
- Locations: Knees; Ankles (Pes equinovarus)
- Onset: Congenital or 1st decade
- Scapular winging: 40%
- Upper motor neuron
- Spastic paraplegia: Some families; Later onset
- Tendon reflexes
- More severe disease: Brisk in legs or arms
- Reduced or absent in legs: Other patients
- Especially at ankles
- Fasciculations: Arms in some patients
- Sensation: Usually normal; Occasional vibratory loss
- Cognitive: Normal
- Course
- Slow progression or Stable
- Many remain ambulatory to 5th decade
- May need wheelchair
- Onset
- Laboratory
- MRI: Muscle
- Pelvis: Gluteus medius & minimus
- Thigh
- Involved: Anterior; Vastus lateralis, intermedius; Sartorius > Rectus femoris
- Spared: Medial adductors & Semitendinosus
- Leg: Posterior calf (Gastrocnemius)
- Similar to DYNC1H1-associated DCSMA
- Electrodiagnostic
- EMG
- Denervation with Reinnervation, Chronic
- No fibrillations
- NCV: Normal
- EMG
- Serum CK: Usually normal; Up to 1100
- Spinal cord pathology
- Motor neurons reduced: Lumbar more than cervical
- Ventral roots: Atrophy
- Muscle pathology
- Chronic denervation
- Type 1 predominance & hypertrophy
- Groups of fast myosin negative muscle fibers
- Fiber size: Varied
- Pseudomyopathic
- MRI: Muscle
- SMALED: Differential diagnosis
- BICD2 variant syndrome: Spastic paraparesis (SPG)
- Epidemiology: 1 family, 4 patients
- Genetics
- Inheritance: Recessive
- Mutations: Especially coiled-coil domain 2; Missense; c.G1823A, p.S608L
- Allelic with: SMALED2
- BICD2 protein
- Clinical
- Onset age: 15 to 18 months
- Spasticity: Gait disorder; Tendon reflexes brisk; Plantar reflex extensor
- Amyotrophy (50%)
- Sensation: Normal
- Cognition: Normal
- Laboratory
- Brain MRI: Normal
- EMG/NCV: Normal
- BICD2 variant syndrome: Arthrogryposis
(SMALED2B)
135
- Epidemiology: 7 sporadic patients
- Genetics
- Inheritance: de novo; Dominant
- Mutations: Gln194Arg, Cys542Trp; Arg694Cys
- Clinical
- Fetal: Hypokinesia
- Skeletal
- Fractures, congenital
- Hip dislocation
- Micrognathia
- Arthrogryposis
- Hypotonia
- Respiratory insufficiency
- Microcephaly
- Cognitive delay
- Muscle: Atrophy
- Course: Early death in some
- Laboratory
- Brain imaging: Perisylvian polymicrogyria; Cerebellar vermis hypoplasia; Corpus callosum thin
- Muscle: Neurogenic atrophy
- EMG: Neurogenic
- BICD2 variant syndrome: "Distal myopathy"
- Epidemiology: 3 families
- Genetics
- Inheritance: Dominant
- Mutations: Arg622Trp
- Clinical
- Onset age
- Weakness
- Symmetric
- Distal legs: Especially foot dorsiflexors
- Laboratory
- Muscle MRI pathology: Thigh anterior & medial; Leg, Anterior > Posterior
- Serum CK: Mildly high
- EMG: Mixed myopathic & Neuropathic
- NCV: Normal Motor & Sensory
- Muscle pathology: Fiber size varied; Endomysial connective tissue increased
Spinal muscular atrophy with Lower limb predominance (SMA-LED)
● Dynein, cytoplasmic 1, heavy chain 1 (DYNC1H1)
- Epidemiology: > 10 patients
- Genetics
- Mutations: Missense; Arg251Cys, I584L, K671E, Y970C; Tail domain & other
- Allelic disorders
- DYNC1H1 protein
- Other dynein related disorder: Congenital SMA with contractures (SMALED2)
- Clinical
- Onset
- Age: Early childhood
- Leg weakness
- Late walking
- Weakness
- Most severe: Quadriceps & Psoas; Hip abduction
- Other: Distal legs; Mild
- Arms: Normal
- Symmetric
- Functional disorders
- Difficulty climbing stairs & rising from chair
- Waddling gait
- Course: Non-progressive
- Wasting: Quadriceps & Distal legs; Small hand muscles
- Tendon reflexes: Reduced at knees; Others normal
- Sensation: Normal
- Contractures: None
- CNS: Learning difficulty in some patients
- Onset
- Laboratory
- Muscle MRI: Thigh
- Early involvement: Vastus lateralis; Sartorius
- Hypertrophy: Adductor longus; Semitendinosus
- Electrophysiology
- EMG: Chronic denervation
- Potentials: Large amplitude, long-duration
- No spontaneous activity
- Distribution: Proximal & Distal Legs; Not paraspinous
- NCV: Normal velocities
- EMG: Chronic denervation
- Muscle biopsy
- Denervation: Small angular muscle fibers
- Type 2 muscle fiber predominance
- Focal inflammation: Perivascular
- Chronic pathology: Muscle fibers small; Endomysium increased
- Muscle MRI: Thigh
- Differential diagnosis
Hereditary Motor Sensory Neuropathy, Proximal (HMSNO; HMSN-P; CMT 2G)
● Trk-fused Gene (TFG)
- Epidemiology: Okinawa, Japan, Brazil, Taiwan (CMT2), Korea families
- Genetics
- TFG protein
- Endoplasmic reticulum exit sites
- Vesicles: Trafficking & Biogenesis
- COPII-mediated export
- Secretory cargoes
- ER → ER-Golgi intermediate compartments
- Inhibition: Slows protein secretion from endoplasmic reticulum (ER); Altered ER morphology
- Full length transcript: Predominantly expressed in neural tissues
- Clinical
- Onset
- Age: Weakness commonly in 5th decade; Range 17 to 50 years
- Cramps: Painful; Onset in 3rd decade
- Fasciculations
- Motor
- Weakness
- Proximal > Distal
- Lower > Upper
- Symmetric
- Cramps
- Fasciculations
- Muscle atrophy
- Course: Progression to severe disability & wheelchair
- Weakness
- Tendon reflexes: Absent
- Sensory
- Loss: Vibration & Joint position > Pain
- Dysesthesias: Distal
- Tremor
- Diabetes Mellitus: 40% with hyperglycemia
- Onset
- Lab
- Hyperlipidemia: 25%
- Hyperglycemia: 30%
- Serum CK: High; 200 to 500
- CNS MRI: Normal
- Electrodiagnostic testing
- CMAP amplitude: Reduced
- SNAP amplitude: Reduced or Absent
- NCV: Velocity normal
- EMG: Denervation: Fasciculations
- Pathology: Spinal cord
- Decreased numbers of anterior horn cells
- Marked loss of myelinated axons in posterior columns
- Neuronal cytoplasmic inclusions: TDP-43, TFG, Optineurin & ubiquitin
- Golgi fragmentation
- Sural nerve
- Myelinated axons: Reduced numbers
- Ultrastructure: Aggregated ER; Mitochondria small
- Muscle
- Fiber size variability
- Internal nucle
- Fiber splitting
- Core-like or targetoid structures
- Grouped atrophy
- Fiber type groups
- Aggregates: TFG, TDP-43, p62
- TFG variant disorder: SPG 57
- Epidemiology: Sudan, Indian & Italian families
- Genetics
- Inheritance: Recessive
- TFG mutations: Arg22Trp, Ile66Thr, Arg106Cys, R106H
- Allelic disorders
- TFG protein
- Mutation
- Inability to self-assemble into oligomeric complex
- Abnormal secretion from ER
- Impaired axon fasciculation after neuronal differentiation
- Mutation
- Clinical
- Onset age: < 2 years
- Spasticity
- Legs > Arms
- Gait: No independent walking
- Tendon reflexes: Increased
- Eye
- Optic atrophy: Reduced visual acuity
- Polyneuropathy
- Distal wasting: Hands & Feet
- Sensation: Normal
- Cognitive: Normal
- Laboratory
- NCV: Polyneuropathy
- Axon loss
- Demyelinating features: NCV reduced; Latencies increased
- Sensory: SNAP amplitudes reduced
- Motor: CMAPs reduced amplitude or Absent
- Brain MRI: Normal
- Nerve pathology: INAD
- CNS: Length dependent corticospinal tract axonopathy
- NCV: Polyneuropathy
- Diferential diagnosis: SPOAN
- TFG variant disorder: SPG, Dominant
200
- Epidemiology: 1 family, 5 patients
- Genetics
- Inheritance: Dominant
- Mutation: Missense; R42Q; Heterozygous
- Mutation effect: Autophagy defect
- Clinical
- Onset age: Early Childhood
- Spasticity
- Legs
- Gait disorder
- Progressive
- Weakness: Legs, Proximal ± Distal
- Laboratory
- Brain MRI: Normal
- NCV: Normal
- EMG: Denervation in legs
- Muscle pathology: Fiber type groups; Grouped atrophy
- TFG variant disorder: CMT2
160
- Epidemiology: Taiwan, Iranian & Italian families
- Genetics
- Mutation: Gly269Val
- Inheritance: Dominant
- Clinical
- Onset age: 28 to 40 years
- Weakness
- Distal
- Arms ≥ Legs
- Cramps
- Sensory
- Loss: Distal; Arms & Legs; Panmodal
- Paresthesias
- Gait ataxia
- Pain: Allodynia; Face
- Tendon reflexes: Reduced in legs
- Course: Progressive; May need walking aids
- Laboratory
- NCV: 44 to 61 M/s; Motor sensory axon loss
- EMG: Chronic denervation
- Nerve pathology: Axon loss, Large myelinated
Spinal Muscular Atrophy, Prenatal onset + Congenital Bone Fractures (SMABF)
● SMABF1
● SMABF2
- Epidemiology
- SMABF1: 16patients
- SMABF2: 13 patients
- Genetics
- Mutations: Stop; Often homozygous
- TRIP4 allelic variant: Congenital muscle disease
- ASCC1 variant: Congenital weakness, Developmental delay, Ulnar epiphysiolysis
- TRIP4 & ASCC1 proteins
- Expressed in embryos: Spinal cord, Brain, Paraspinal ganglia (Dorsal & Sympathetic), Thyroid & Submandibular glands
- ASC-1 transcriptional cointegrator complex
- Subunits: TRIP4; ASCC1; ASCC2; ASCC3
- Ribonucleoprotein complex
- Participates in: Transcriptional coactivation, RNA processing events
- ASC-1 binding protein: Cysteine and glycine rich protein 1 (CSRP1)
- Role in late myogenesis
- Expressed at low level in muscles, especially axial
- TRIP4 & ASCC1 location: Nucleus
- Clinical
- Genetic correlations
- ASCC1 mutations: More fractures; Shorter survival
- Pregnancy: Reduced fetal movements
- Motor
- Hypotonia
- Respiratory difficulty
- Diaphragm eventration
- Pulmonary hypoplasia
- Weakness
- Muscle mass: Decreased
- Tendon reflexes: Absent
- CNS: Global developmental delay
- Bone
- Generalized osteopenia
- Congenital fractures: Multiple; Long bones
- Joints: Arthrogryposis
- Multiple contractures: Proximal & Distal
- Congenital heart defects
- Secundum atrial septal defect
- Patent ductus arteriosus
- Cardiomyopathy
- Other: Some patients
- Face dysmorphism
- Microretrognathia,
- Hypertelorism
- High-arched palate
- Hypertrichosis
- Face dysmorphism
- Course: Death at 2 to 16 months
- Genetic correlations
- Laboratory
- EMG
- Neurogenic
- Muscle: No contraction to stimulation
- Pathology
- Muscle
- Fiber size: Atrophy; Variable
- Type I fibers: Larger; Clustered
- Fiber immaturity: Immature myosin (Developmental)
- Western blot: TRIP4 with reduced size, Upregulation of splice isoform
- Sural nerve: Loss of unmyelinated axons; Collagen pockets
- Spinal cord
- Anterior horn cells: Loss; Apoptosis
- Astrogliosis
- Muscle
- Brain MRI: Cortical gyration abnormal
- EMG
- TRIP4 variant: Congenital muscle disease (MDCDC)
132
- Nosology: Congenital muscular dystrophy, Davignon-Chauveau type
- Epidemiology: 6 families
- Genetics
- Inheritance: Recessive
- Mutations: c.G950A, p.W297*; Stop, Splice, Missense (Milder phenotype)
- Clinical
- Onset ages: Birth to adult
- Motor
- Hypotonia: Neonatal; Axial
- Respiratory failure
- Feeding difficulties
- Motor development: Delayed
- Weakness: Severe; Especially Proximal & Trunk
- Skeletal: Some
- Joint hyperlaxity
- Scoliosis: May be severe
- Rigid spine
- Contractures: Mild or None
- Dysmorphic: Flat face; Thick neck
- Skin
- Hyperelasticity
- Dry with scratch lesions
- Follicular hyperkeratosis
- Xerosis
- Cardiac: Dilated; Some patients
- Ophthalmoplegia: 2 patients
- Course
- Death in 1st years: Some
- Late: Non-walking
- Often stable
- Laboratory
- Serum CK: Normal or Mildly high
- EMG: Myopathic
- Muscle CT: Preservation of leg adductors
- Muscle MRI: Posterior thigh involvement with semi-tendinosus sparing
- Muscle biopsy
Pontocerebellar hypoplasia with Spinal muscular atrophy (PCH1A)
● Vaccinia-related kinase 1 (VRK1)
- Epidemiology: 3 families
- Genetics
- Mutations: Nonsense or Missense 68
- Allelic with: Motor Neuron Disease, Distal ± Sensory loss
- VRK1 protein
- Serine/Threonine kinases
- Phosphorylates p53 & CREB
- Localization: Nuclear + Some cytoplasm & cell membrane
- Role in: Nuclear envelope formation
- Expression: Ubiquitous including brain
- Clinical
- Onset
- Age: 1st decade
- Severe cases: Prenatal or Birth; Death within months
- Milder cases: < 6 months
- Fetal movements: Reduced
- Hypotonia
- Feeding difficulty
- Age: 1st decade
- Motor
- Hypotonia
- Weakness
- Early: Reduced movements
- Diffuse
- Feeding disorders
- Progressive: Walking achieved in some then lost
- Milestones: Delay & Decline
- Polyneuropathy: Distal, Symmetric
- Tendon reflexes: Reduced or Increased
- CNS
- Cerebellar: Ataxia; Nystagmus
- Psychomotor retardation: Some patients
- Skeletal
- Contractures: Variable;
- Arthrogryposis: Severe cases
- Mild: Later onset patients
- Scoliosis
- Contractures: Variable;
- Course: Survial ranges from months to 12 years
- Onset
- Laboratory
- Serum CK: Normal
- MRI
- Cerebellar hypoplasia or absence, including vermis
- Pontine hypoplasia: Severe cases
- Microcephaly
- EMG: Neurogenic
- NCV: Axonal sensory-motor neuropathy
- Motor & Sensory velocities: Normal or Mildly slowed
- CMAP & SNAP amplitudes: Small
- Pathology
- Muscle: Denervation; Fiber type grouping; Grouped atrophy
- Nerve: Loss of large myelinated & small unmyelinated axons in sural nerve
- CNS
- Spinal cord: Motor neuron loss
- Cerebellum: Hypotrophic; Absent dentate nucleus; Other structural defects
- Brainstem: Hypotrophic; Posterior fossa cysts
- Basal ganglia: Neuronal loss
- Cortical atrophy
- VRK1 variant syndrome: Motor Neuron Disease, Distal ± Sensory loss (HMNR10)
124
- Epidemiology: 7 patients
- Genetics
- Inheritance: Recessive
- Mutations
- Missense
- Compound heterozygous
- V236M; p.H119R (c.356A>G); p.R321C (c.961C>T); R358*; G135R; L195V
- Clinical
- Onset age: 3 to 31 years
- Distribution
- Distal
- Legs (especially posterior) > Arms: Ankles & Toes; Hands
- Symmetric
- Respiratory
- Gait disorder
- Course: Slow progression over years
- Distribution
- Muscle atrophy: Distal legs, posterior
- Tendon reflexes: Brisk, except absent at ankles
- Sensory loss: Small fiber modalities in legs
- Skeletal: Pes cavus; Hammer toes
- Onset age: 3 to 31 years
- Laboratory
- Serum CK: High (347 to 3,400) or Normal
- NCV
- Motor: CMAPs small
- Sensory: Normal
- EMG
- Distal legs: Active denervation (Fibrillations)
- Arms & Proximal legs: Chronic denervation with reinnervation (Large motor units)
- Muscle biopsy: Small angular muscle fibers; Nuclear clumps
- Brain MRI: Normal
Pontocerebellar hypoplasia + Spinal Muscular Atrophy (PCH1B)
● Exosome component 3 (EXOSC3)
- Epidemiology: 50 patients; 31% of PCH1
- Genetics
- Mutations: Missense (Most common); Deletion; Splice-site
- Common mutation: Asp132Ala; Homozygous associated with milder phenotype
- Altered reading frame mutations: More severe; Death in childhood
- No homozygous nulls reported
- Allelic disorders
- EXOSC3 protein
- RNA exosome core component
- Cellular locations: Cytoplasm; Nucleus; Nucleolus
- Exosomes
- EXOSC1�EXOSC3: Cap of exosomal ring for RNA recognition & binding
- EXOSC4�EXOSC9: Form core, a hexamer channel through which RNA passes
- Exosome-related disorders
- Cerebellar
- PCH: EXOSC1
- PCH1B: EXOSC3
- CABAC: EXOSC5
- SMA + Cerebellar hypoplasia (PCH1C) - EXOSC8
- Cerebellar atrophy + Spinal Motor Neuropathy: EXOSC9
- Spinal motor neuropathy: RBM7
- Immune: PM/Scl (EXOSC9 & EXOSC10) autoantibodies
- Cerebellar
- Clinical
- Onset: Congenital
- Motor neuron: Muscle wasting
- Contractures: Distal
- Cerebellar
- Oculomotor apraxia
- Microcephaly: Progressive
- Developmental delay: Global; Severe
- Laboratory
- MRI: Cerebellar atrophy; Brainstem & Cortex small
- EMG: Neurogenic; Large motor units
- NCV
- CMAP amplitude: Small
- Sensory responses: Normal
- Autopsy
- Cerebellum: Neuron loss (Purkinje & Granule)
- Spinal cord: Motor neuron loss
- EXOSC3 Variant syndrome: Spastic paraplegia, Early onset & Complicated
104
- Epidemiology: Bangladesh-Italian & Arab-Israel families
- Genetics
- Inheritance: Recessive
- EXOSC3 mutations: V80F; D132A; G191C
- Clinical
- Onset age: 1 to 4 years
- Spasticity
- Legs > Arms
- Gait disorder
- Tendon reflexes: Brisk
- Plantar response: Extensor
- Cognitive impairment: Mild
- Speech disorder
- Learning dificulty
- Cerebellar:
- Dysarthria
- Nystagmus
- Intention tremor & Dysmetria
- Motor: 1 family
- Distal amyotrophy
- Tongue atrophy
- Fasciculations
- Eye: Strabismus (50%)
- Skeletal: Adducted thumbs, Talipes valgus; Short stature (50%); Normal head circumference
- Course: May be progressive after 1st decade
- Laboratory
- EMG: Distal denervation
- Nerve conduction: Velocities normal
- Brain MRI: Cerebellar atrophy
- Muscle biopsy: Type 2 predominance; Varied muscle fiber size
- SSEP: Delayed central conduction times
Cerebellar atrophy + Spinal Motor Neuropathy (PCH1D)
● Exosome component 9 (EXOSC9)
- Nosology: Ponto-Cerebellar Hypoplasia 1D
- Epidemiology: 4 patients
- Genetics
- Mutations: Stop; c.481C>T, c.41T>C (Milder phenotype)
- EXOSC9 protein
- Exosome core protein
- Part of hexamer channel through which RNA passes
- PM/Scl antigen
- Other exosome disorders
- Clinical
- Onset age: Prenatal or Congenital
- Prenatal: Reduced movements; Growth retardation
- Infant: Poor head control
- Motor
- Delayed development
- Hypotonia
- Weakness: Generalized; Proximal & Distal; Respiratory
- Fasciculations: Tongue; Limbs
- Sensation: Normal
- Tendon reflexes: Reduced or Present
- Skeletal
- Joint contractures & Arthrogryposis
- Neonatal fractures
- Face: Dysmorphism
- Growth delay
- Seizures: 1 patient
- Course: Progressive
- Laboratory
- EMG: Motor neuropathy; Fasciculations
- NCV: Motor axon loss
- Muscle biopsy: Fiber type groups; Grouped atrophy; Type 1 fibers large
- Brain MRI: Cerebellar atrophy, progressive
- Serum CK: Borderline
Ponto-Cerebellar Hypoplasia + Spinal Motor Atrophy & Arthrogryposis (PCH)
● Kinesin Family Member 26B (KIF26B)
- Epidemiology: 1 Indian patient
- Genetics
- Mutation: Missense, Gly546Ser; Heterozygous
- KIF26B protein
- Kinesin, Unconventional
- Intracellular motor protein
- Cell polarity of migrating endothelial cells during angiogenesis
- Clinical
- Onset age: Congenital
- Face dysmorphism: Microcephaly
- Skeletal: Arthrogryposis; Camptodactyly; Dislocated joints
- Respiratory failure
- Course: Death at 7 months
- Laboratory
- Brain MRI: Microcephaly; Ponto-Cerebellar Atrophy; Spinal cord thin; Atrophy, progressive
- Muscle biopsy: Atrophic fibers
- Nerve biopsy: Reduced numbers of myelinated axons
- EMG: Motor neuron loss
Lower motor neuron syndrome, Childhood onset (HMNR4; DSMA4)
● Pleckstrin homology domain-containing protein, Family G, Member 5 (PLEKHG5)
- Epidemiology: Several families
- Genetics
- Mutations
- Location: C-terminal region common
- Pro630His; Pro27Ter; Val455Gly; Gln550X; Met557Leu; Phe647Se; c.2057delT, c.2752_2753delGG; Thr686Met; Pro707His
- Allelic with: CMT-RIC
- Same mutations may produce DSMA4, CMT-RIC or Overlap phenotype
- Mutations
- PLEKHG5 protein
- Cytoplasmic
- Distribution: Ubiquitous; High in peripheral nerve (endoneurium), spinal cord & brain
- Function: Nuclear Factor κB�Activator
- Mutant protein
- Reduced levels in cells
- Loss of NFκB-activating function
- Aggregates
- Small GTPase signalling pathways: Dysregulation
- Autophagy disturbed
- Proteins sharing PH or PH/RhoGEF domain: Dynamin 2; Alsin
- Other guanine nucleotide exchange factor (GEF) disorders
- Clinical
- Onset
- Age: 2 to 25 years
- Weakness
- Weakness
- Distribution: Generalized; Proximal ≥ Distal; Symmetric
- Respiratory: With disease progression
- Scapular winging
- Face & Cranial nerves (Bulbar): Normal
- Course
- Progressive over a decade to severe disability
- Milder phenotype in some patients
- Tendon reflexes: Reduced or Absent
- Contractures: Hips; Elbows; Hands
- Spine: Hyperlordosis; Scoliosis
- Intelligence: Normal
- Sensory: Normal or Mild loss
- No upper motor neuron signs
- Onset
- Laboratory
- EMG: Denervation; Motor units large
- NCV
- Motor: Normal or Slow velocities; CMAPs may become small
- Sensory: Normal or Mildly slow
- Serum CK: Mildly high
- Muscle biopsy: Denervation
- Muscle MRI: Fatty replacement in glutei & thighs > legs
- Variant syndrome: Charcot-Marie-Tooth disease, Recessive Intermediate C (CMTRIC)
- Epidemiology: Korean, Portuguese & Moroccan families
- PLEKHG5 Genetics
- Inheritance: Recessive
- Mutations
- Compound heterozygous or Homozygous
- Stop; Deletion; Duplication (7 bp); Missense
- Allelic with: DSMA4
- PLEKHG5 protein
- Clinical
- Onset age: 1st to 5th decades
- Weakness
- Distal
- Proximal With disease progression; Legs & Arms
- Arms & Legs
- Course: Progressive
- Tendon reflexes: Reduced or Absent
- Sensory loss
- Distal
- Legs > Arms
- Modalities: Large & Small axon
- Varied severity
- Skeletal: Pes cavus; Spine deformity in 1 family
- Laboratory
- Serum CK: May be mildly increased
- NCV
- Motor: Intermediate slowing (24 to 39 m/s in median nerve)
- SNAPs: Absent or Reduced amplitude
- EMG: Denervation, Distal
- Muscle MRI: Fatty replacement, especially anterior & lateral distal legs
- Normal: VER; BAER
- Nerve biopsy
- Axon loss: Large > Small; Severe
- Regenerating axon clusters
- Hypomyelination
- PLEKHG5: Absent from axons; Present in Schwann cell nuclei
- Muscle: Neurogenic
Distal Motor Neuropathy with Young Adult Onset (HMNR5; DSMA5; dHMN)
● DNAJ/HSP40 Homolog, subfamily B, Member 2 (DNAJB2; HSJ1)
- Epidemiology: 5 families
- Genetics
- Mutations
- Homozygous
- Splice site (c.229+1G>A, c.352+1G>A); Deletion (c.310delC, 3.8-kb deletion); c184C>T;
- Recessive mutations: Loss-of-function
- Allelic disorders
- Other DNAJ disorders
- Mutations
- DNAJB2 (HSJ1) Protein
- Molecular Co-Chaperones
- HSP40/DNAJ family
- Cellular location: Nuclear & Cytoplasmic
- Isoforms
- 2 J-domain co-chaperones: Varied C-termini
- HSJ1b
- Membrane anchored
- Cytoplasmic face of endoplasmic reticulum (ER) by C-terminal prenylation
- Perinuclear
- Promotes proteasomal degradation of ER-located proteins: Via ER-associated degradation (ERAD) pathway
- HSJ1a
- Cytosolic or Nuclear
- Counteracts TDP-43 aggregation: Promotes HSPA-mediated refolding
- Expression: Strong in neurons; Post-synaptic NMJs in muscle
- Protects ubiquitylated clients against activity of ubiquitin hydrolases
- Mediates degradation of HSPA client proteins: Via ubiquitin�proteasome system
- J-Domain
- Stimulates ATPase activity of HSPA (Hsp70) chaperones
- Enables chaperone cycle: Alternating binding & release of client (substrate) proteins
- Has client-specific refolding activity
- May reduce aggregate formation
- Interactions: STUB1
- Mutation: Causes truncated protein
- Clinical
- Onset
- Age: 1st to 4th decade
- Foot drop
- Gait disorder
- Weakness
- Distal: Severe in legs
- Legs > Hands
- Bulbar & Respiratory: Later in course
- Course: Progressive to wheelchair
- Sensory: Normal early; Loss later in course
- Tendon reflexes: Absent
- CNS: Parkinson disease in some patients
- Edema: Legs
- Onset
- Laboratory
- Electrodiagnostic
- CMAP amplitudes: Reduced, especially in legs
- NCV: Normal velocity
- SNAPs: Normal
- EMG: Denervation, Distal legs
- Muscle: May have rimmed vacuoles
- Electrodiagnostic
- DNAJB2 (HSJ1) variant: AR-CMT 2
117
- Epidemiology: 4 families
- Genetics
- Inheritance: Recessive
- Mutations: Homozygous; ; Most stop, Some missense; Tyr5Cys, c.145delG, c.310delC, c.619-1G>A
- Clinical
- Onset ages: 2nd or 3rd decade
- Weakness
- Foot & toe dorsiflexion: Severe
- Hands & Arms
- Hip & knee flexors
- Gait: Unstable
- Sensory: Leg paresthesias
- Tendon reflexes: Absent in legs
- Some patients/families
- Parkinsonism
- Hearing loss: May be severe
- Course: Progression to severe weakness
- Laboratory
- NCV: Axonal motor & sensory length-dependent neuropathy
- EMG: Denervation, Distal legs
- Nerve pathology: Loss of myelinated axons
- Muscle CT: Fat replacement in leg & thigh muscles
- Skin: Loss of DNAJB2 in axons
- DNAJB2 variant: Neuromyopathy
195
- Epidemiology: 1 family; Son, sib & possibly mother
- Genetics
- Inheritance: Dominant
- Mutation: c.832T>G (p.*278Glyext*83)
- Mutation effects
- Abolishes stop codon on DNAJB2a isoform
- Dominant negative
- Clinical
- Onset age: 5th decade
- Gait disorder: Imbalance; Slow; Ataxia
- Weakness: Legs, distal; Fatigue
- Muscle atrophy: Distal legs
- Sensory loss: Vibration, distal legs; Joint position
- Paresthesias: Distal legs
- Tendon reflexes: Reduced or absent in legs
- CNS: Normal
- Course: Slow progression
- Laboratory
- Serum CK: 7x increased
- Head & Spine MRI: Normal
- Muscle MRI: Myopathy & Neuropathy; Fatty replacement
- NCV: Axon loss, sensory & motor; Long F-wave & distal latencies; Velocities normal
- EMG: Chronic neurogenic; No spontaneous activity
- Muscle pathology: Fiber type groups; Grouped atrophy; Internal nuclei; Vacuoles
- Nerve pathology: Axon loss
Lower motor neuron syndrome with late-adult onset (SMAJ; LOSMoN)
● CHCHD10
- Nosology: Spinal muscular atrophy, Jokela type
- Epidemiology
- 17 Finnish families
- Eastern Finland (Northern Karelia) prevalence: 12:100,000
- Genetics
- Mutation: G66V
- Allelic disorders
- CHCHD10 protein: Mitochondrial
- Clinical
- Onset
- Age: 30 to 73 years
- Weakness, Cramps or Fasciculations
- Weakness
- Proximal > Distal + Abdomen
- May be: Asymmetric or Distal
- Cramps: Proximal > Distal; Painful
- Fasciculations: Distal & Proximal
- Tendon reflexes: Absent diffusely or in legs
- Other occasional features
- Myalgias (50%)
- Drop attacks: Body or head
- Tremor: Associated with hand fasciculations
- Ataxia
- Pes cavus: Mild
- Vibration sense reduction (50%): Distal legs; Asymmetric
- Course
- Progression: Slow
- Most retain some ambulation
- Onset
- Laboratory
- Serum CK: Normal to 8x High
- NCV
- Normal velocities
- CMAP amplitudes: Normal or Reduced
- SNAPs: Normal or Mildly small
- EMG: Denervation, Distal & Proximal
- Motor unit potentials: Long duration; Large amplitude
- Fibrillations
- Fasciculations
- Complex repetitive discharges
- Recruitment: Reduced
- MRI
- Posterior lower leg muscles abnormal
- Especially medial gastrocnemius
- Muscle biopsy
128
- Fiber type grouping
- Grouped atrophy
- Pyknotic nuclear clumps
- Type 2 fibers: Small
- Largest muscle fibers: Hypertrophy
- Neonatal myosin heavy chain (MHCn): 3%-50% (median 15%) of muscle fibers
- Vacuoles
- Frequency: 60% of biopsies
- Fibers also stain for LC3, p62, SMI-31 & TDP-43
- More common with longer disease duration
Proximal Spinal Muscular Atrophy with Progressive Myoclonic Epilepsy (SMA-PME; SMAPME)
● N-Acylsphingosine Amidohydrolase 1 (ASAH1; Acid ceramidase; Acylsphingosine deacylase)
- Epidemiology: 50 patients
- Genetics
- Mutations
- Types: Missense, Thr42Met, Thr42 Ala (Milder phenotype); Deletion, Small or whole gene
- Homozygous
- Allelic disorders
- Farber Lipogranulomatosis: Very low ASASH1 levels
- Progressive adult-onset brachydactyly due to osteolysis
- SMA without epilepsy
- Mutations
- ASAH1 protein
- Localization: Lysosome
- Maintains intralysosomal ceramide homeostasis
- Cleaves Ceramide into Sphingosine & free fatty acids at acid pH

- Clinical
- Onset age: 1 to 30 years; Mean 6 years
- Weakness
- Proximal
- Legs, then Arms; Face, mild
- Respiratory: Later in disease course
- Tongue fasciculations
- Tendon reflexes: Absent or Present
- Tremor: Postural
- Seizures
- Myoclonus epilepsy
- With more severe phenotypes
- No contractures
- Skin: Normal
- Course
- Progressive: To severe disability
- Death: Some patients in 2nd decade
- Laboratory
- EMG: Chronic denervation
- NCV: CMAPS small; SNAPs normal
- EEG
- Epilepsy: Subcortical myoclonic epileptiform activity, sensitive to hyperventilation
- May be normal
- Serum CK: Normal
- Muscle biopsy: Denervation & Reinnervation
- Variant syndrome: Farber Lipogranulomatosis
- Genetics
- ASAH1 mutations: Cause severe reduction in Acid ceramidase activity (< 10%)
- Inheritance: Recessive
- Clinical
- Skin: Lipogranulomata, subcutaneous; Periarticular subcutaneous nodules
- Joints: Contractures; Pains
- Hoarse voice
- CNS: Mental retardation; Progressive deterioration
- Lower motor neuron involvement (15%)
- Hypotonia
- Muscle atrophy
- Respiratory insufficiency
- EMG: Denervation
- Systemic
- Hepatomegaly
- Splenomegaly
- Eyes: Macular cherry red spots
- Death: < 2 years
- Laboratory
- NCV: Slowing in some patients
- Nerve pathology
- Schwann cells, myelinating: Banana bodies
- Genetics
Spinal Muscular Atrophy with Hypomyelination & Cerebellar Hypoplasia (PCH1C)
● Exosome component 8 (EXOSC8)
- Epidemiology: 3 families & 22 patients, Hungarian & Arab-Palestinian
- Genetics
- Mutations: Missense; Ala2Val, Ser272Thr
- Other exosome disorder: PCH1B - EXOSC3
- EXOSC8 protein
- Exosome component
- Multi-protein complex
- Functions
- 3'->5' exoribonuclease activity
- Participates in many cellular RNA processing & degradation events
- Degradation of AU-rich element (ARE) containing mRNAs
- Immune disorders: Exosome = PM/Scl target of autoantibodies
- EXOSC8
- Part of central hexamer channel of exome
- Non-catalytic component
- Cell location: Nucleus & Cytoplasm
- Exosome component
- Clinical
- Onset age: Infant; 2 to 4 months
- Failure to thrive
- Weakness: Severe
- Spasticity
- Psychomotor retardation
- Vision: Impaired
- Hearing: Impaired
- Deterioration: Triggered by inter-current infections
- Death: Respiratory failure < 20 months
- Laboratory
- CNS
- Cerebellar hypoplasia
- Hypomyelination: Especially lateral descending tracts
- Muscle
- Fiber size: Varied
- Cytochrome oxidase: Some negative fibers
- Mitochondrial oxidative enzymes: Complex I & IV reduced
- Peripheral nerve: Normal myelin
Encephalopathy with Distal Spinal Muscular Atrophy, Early-onset, Progressive (PEAMO)
● Tubulin-specific chaperone E (TBCE)
- Epidemiology: 4 families, 6 patients
- Genetics
- Mutations: c.463Tgt;A (p.Ile155Asn) homozygous or + Stop
- Allelic disorders
- Kenny-Caffey syndrome
- Sanjad-Sakati syndrome
- Clinical features
- Congenital hypoparathyroidism
- Mental retardation
- Face dysmorphism
- Growth failure
- Kenny-Caffey syndrome
- TBCE protein function
- Tubulin-specific chaperones
- Folding of α-tubulin
- Formation of α-β-tubulin heterodimers
- Heterodimer polymerization into microtubules
- See: SMA + TBCD mutations
- Clinical
- Onset age: Neonatal to 14 months
- Hypotonia
- Developmental delay
- Speech: Absent or Dysarthria
- Cognition: Moderate to Severe involvement
- Weakness & Wasting
- Distal
- Legs & Arms
- Spasticity: Tetraparesis
- Ataxia (60%)
- Optic atrophy (40%)
- Scoliosis
- Course: Progressive
- Laboratory
- Brain MRI
- Cerebellar atrophy
- Corpus callosum hypoplastic
- Similar to NBIA
- Muscle pathology: Small angular fibers; Fiber type grouping
- EMG: Motor units large; Distal fibrillations; No fasciculations
- NCV: CMAP amplitudes reduced; Conduction velocities normal
- VEPs & BAERs: Delayed
- Calcium/Phosphate metabolism: Normal
- Brain MRI
- Mouse model: pmn
- Genetics
- TBCE Mutation: Trp524Gly
- Gene location: Mouse chromosome 13
- Loss of motor neurons & myelinated axons
- CNS
- Tracts - Fasciculus gracilis; Rubrospinal; Reticulospinal
- Normal: Corticospinal tract
- Pathological pattern: Dying back
- Death: 6 weeks
- Genetics
Spinal Muscular Atrophy, Early-onset with Neurodegeneration 136
● Tubulin-specific chaperone D (TBCD)
- Epidemiology: 1 family 2 patients
- Genetics
- Mutation: Homozygous; R942Q
- Allelic disorder
- Encephalopathy, progressive, early-onset, with Brain atrophy & Thin corpus callosum
: Has motor neuron loss
- Encephalopathy, progressive, early-onset, with Brain atrophy & Thin corpus callosum
- TBCD protein
- Microtubule assembly
- Peripheral nerve: Axon & Dendrite maintenance
- See: dHMN + TBCE mutations
- Clinical
- Hypotonia
- Developmental disorder
- Weakness: Diffuse; Prfoximal > Distal; Face; Respiratory
- Fasciculations: Tongue
- Microcephaly
- Seizures
- Psychomotor retardation
- Vision loss: Optic atrophy
- Laboratory
- Serum CK: Normal
- Brain MRI: Cerebral atrophy
- NCV: CMAP amplitudes reduced; Velocities reduced
- Muscle: Grouped atrophy; Hypertrophy of largets muscle fibers
- Sural nerve: Normal
Motor Neuropathy & Intellectual Disability (TBCK Encephaloneuronopathy; IHPRF3)
● TBC1 Domain-containing Kinase K (TBCK)
- Epidemiology
- > 10 patients
- Common: Puerto Rican children of Boricua descent (Arg126X); Carrier rate 1:45
- Genetics
- Mutations: c.1652T>C (p.L551P); Arg126X
- Allelic disorder
- Hypotonia, infantile, with Psychomotor retardation & Characteristic facies 3
: > 100 patients; mtDNA reduced
- Heterozygotes: Deformities, Toe & Foot; Apraxia; Peripheral neuropathy
- Hypotonia, infantile, with Psychomotor retardation & Characteristic facies 3
- TBCK protein
- Protein kinase
- Associates
- Mitotic apparatus
- Proteins: PPP1R21; c12orf4; CRYZL1
- Vescicular organelles in neurons
- Ferry complex: Transport mRNAs
- Regulates: Cell size, Cell proliferation, MTOR
signaling
- Axonal mRNA trafficking
- Clinical
- Onset age: Child
- Hypotonia
- Weakness
- Diffuse: Distal > Proximal
- Respiratory insufficiency by teenage years
- Rarely sit
- Tendon reflexes: Absent
- Face
- Ptosis
- Hypotelorism
- Coarse
- Macroglossia
- CNS
- Autism
- Speech & Cognitive delay
- Seizures: Focal & Generalized
- ? Exacerbation by ketogenic diet
- Course: Progressive
- Systemic
- Osteoporosis
- Renal: Calculi; Urinary retention
- Hypothermia: Intermittent to 33°C
- Brachymelia
- Course: Progressive
- Laboratory
- Motor axon loss
- Dyslipidemia: HIgh cholesterol or triglycerides
- TBCK- fibroblasts: Increased LC3+ autophagosomes
- Urine: Free oligosaccharide profiles
- Brain MRI: Leukoencephalopathy; Atrophy
- NCV: Motor neuronopathy
- CMAPs: Small amplitude
- SNAPs: Generally preserved
- Velocities: Normal
- EMG: Chronic partial denervation; Myokymia in 1 patient; Some myopsthy in teenage years
- Muscle ultrasound: Atrophic; Dense; Fasciculating movements
- Muscle & Nerve biopsies: Non-diagnostic; COX deficiency
MTDPS18: Spinal Muscular Atrophy-like disorder
● Solute carrier family 25 (Mitochondrial oxodicarboxylate carrier), Member 21 (SLC25A21)
- Epidemiology: 1 Pakistani patient
- Genetics
- Mutation: Lys232Arg; Homozygous
- SLC25A21 protein
- Mitochondrial
- Oxodicarboxylate carrier
- Clinical
- Onset age: 3 years
- Weakness
- Distal then Proximal
- Arms & Legs
- Face: Mild
- Respiratory
- Gait disorder
- Muscle atrophy
- Exacerbations: Associated with fever
- Tendon reflexes: Brisk
- Scoliosis
- Course: Progressive; Wheelchair at 15 years
- Laboratory
- Anemia: Microcytic
- NCV: Motor axon loss
- Nerve: Axon loss
- Muscle
- Groups of
- Atrophic & Hypertrophied muscle fibers
- Fiber types
- Type 1 predominance
- Mitochondrial
- COX- muscle fibers
- Complex I & IV activities: Reduced
- mtDNA depletion
- Groups of
- Brain MRI: Normal
- CSF: Lactate mildly high
- Urine: Excretion of 3-Hydroxyisovaleric & Glutaric acid
Motor Neuropathy, Distal 185
● Neuronal cell adhesion molecule (NRCAM)
- Epidemiology: 2 families
- Genetics
- Mutations: Ser134Pro; Gln25X
- Allelic disorder: Neurodevelopmental disorder with Developmental delay, Hypotonia, Spasticity
- NRCAM protein
- Location: Neurons & Glial cells in Ventral midline
- Adhesion molecule
- Immunoglobulin (Ig)-CAM
- L1 subgroup
- Neural specific
- Nervous system development
- Axon pathfinding
- Clinical
- Onset age: 2nd decade
- Weakness
- Distal
- Legs > Arms
- Skeletal: Scoliosis; Pes cavus; Hammer toes
- Laboratory
- EMG: Denervation, distal; Myopathic, proximal
- NCV: CMAPs small; SNAPs normal
- Serum CK: 2x to 3x high
- NRCAM variant: Neurodevelopmental disorder with Developmental delay, Hypotonia, Spasticity (NEDNMS)
- Epidemiology: 8 patients
- Genetics
- Mutations: Missense & Stop
- Inheritance: Recessive
- Clinical
- Onset age: Birth
- Developmental delay
- Cognitive disorder
- Hypotonia (50%)
- Spasticity (50%)
- Ataxia
- Skeletal: Dysmorphism; Microcephaly; Scoliosis; Hip dislocation
- Cryptorchidism
- Respiratory: O2 supplementation
- Eye: Strabisums (30%); Optic atrophy
- Hearing loss
- Laboratory
- Brain imaging: Ventricles large; Myelination Δ
- NCV: Polyneuropathy
Motor Neuropathy 194
● Barrier-to-Autointegration Factor 1 (BANF1)
- Epidemiology: 1 patient
- Genetics
- Mutation: Missense; Gly16Arg
- Allelic disorder: Néstor�Guillermo progeria
: Ala12Thr mutation
- BANF1 protein
- Locations: Nuclear lamina; Phosphorylated forms in cytoplasm
- Regulation of gene expression, cell cycle progression, nuclear integrity
- DNA binding: Bridges double-stranded DNA into ordered nucleoprotein complex
- Loss: Absence of mitotic cells; Aberrant nuclear lamina structure
- Interacts with: Histones; Chromatin modifying proteins; Lamin A/C; Emerin; PARP1 in response to oxidative stress
- Clinical
- Onset age: 3 years
- Gait disorder
- Weakness
- Diffuse: Complete foot drop
- Legs > Arms
- Respiratory
- Tendon reflexes: Reduced at ankles
- Sensation: Cold reduced at toes
- Intention tremor: Mild
- Course: Slow progression
- Laboratory
- NCV: Normal velocities
- EMG: Denervation, diffuse
SMA: Severe infantile
● Recessive
Scapuloperoneal syndromes (? Dominant; Sporadic)
ALS: Hereditary
|
|
Protein Pathways Protein Quality SOD1 VCP OPTN TBK1 UBQLN2 p62 NEK1 FIG4 CCNF CYLD Cytoskeletal PFN1 DCTN1 TUBA4A EPHA4 KIF5A ANXA11 PRPH RNA pathology c9orf72 TDP43 FUS TAF15 ELP3 ANG NEK1 TIA1 ATXN2 Vesicles Alsin CHMP2B VAPB Lipids SPTLC1 SPTLC2 |
General: Hereditary vs Sporadic ALS
| Feature | Hereditary ALS | Sporadic ALS |
| Males:Females | 1:1 | 1.7:1 |
| Disease Duration | Bimodal < 2 & > 5 years |
Unimodal 3 to 4 years |
| % of ALS cases | 5% to 15% | ~90% |
| Onset | ||
| Age distribution | More younger | More older |
| Mean age | 46 years | 56 to 63 years |
| Juvenile | ALS 2, 4, 5, 6 | Rare |
| Bulbar features | 25% c9orf72 40% ALS1 10% |
15% |
| Legs | Common | Occasional |
| LMN predominant | ALS 1, 6, 17 | PMA |
ALS SYNDROMES: DOMINANT
- Amyotrophic Lateral Sclerosis 1 (ALS 1)
● Superoxide Dismutase 1 (SOD1); Chromosome 21q22.11; Usually Dominant
Clinical features
General
Specific syndromes
Mutations
Location
Functional aspects
Specific correlations
Pathology
Population statistics
SOD1 other
SOD1 protein
Variants
Canine disorder
SOD1 deficiency
From: M WeissALS-SOD1 (A4V): Tongue atrophy
- Population statistics
- Frequency of SOD mutations in ALS syndromes
- 12% to 23% of Familial ALS
- 1% to 3% of Sporadic ALS
- 1% to 3% of all ALS
- China: Most common F-ALS gene (59%)
- Penetrance: Overall 85% by age 85
- Frequency of SOD mutations in ALS syndromes
- Genetics
- Inheritance: Dominant; de novo; Recessive
- Mutations: Location & Character
- > 200 different disease related mutations identified
- 55 likely pathogenic: Based on linkage, population studies, or hotspot location
- Locations
- Usually in exons 1, 2, 4 & 5
- Few in exon 3: 6 exon 3 mutations identified
- 6 different mutations identified at codon 93 (Gly93) in exon 4
- Most common mutations: D90A; A4V; I113T; L144F; G41D; E100K
- Hot spots: A4; C6; N86; G93; D101; L126; N139;L144
- Mutations: Common features
- Heterozygous
- Missense
- Point
- Dominant inheritance with complete penetrance
-
A4V; G37R; L38V; G41S; H43R; H46R; D76V; L84F; L84V;
N86K; E100G; D101F; I104F; G108V; C111Y; I112M; G114A;
L126X; G127X; G141E; L144F; V148G; V148I - China 181: V47A, H46R, C111Y, G147D
- Finland: D90A
- Dominant de novo
179
- Slow progression: Gly38Arg; His47Arg
- Other: His81Arg; Ala90Thr; Asp91Val; Asp102Asn (Rapid progression)
- Other types
- Stop codon
183
- 17 nonsense mutations identified
- Nonsense mutations producing disease
- Locations: Outside AA 50 to 100
- Escape nonsense mediated decay
- Produce polypeptide length of 121 tp 156 aa (Normal 153)
- Exon 5 not needed for toxic action of mutated SOD
- Intron: IVS4AS, A-G, -11 (11 bases upstream from intron-junction of exon 5)
- Deletion
- Intronic: Altered splice site; 3' untranslated region
- Recessive
- D90A; ? D96N
- Dose effect (Homozygous more severe): L84F, N86S, L126S
- Incomplete penetrance 76
- Stop codon
183
- External link: ALS mutation database
- Mutations: Functional aspects
- Most mutations affect subunit protein folding or dimer contact.
- Mutations may alter some Cu binding sites
- Mutant SOD enzyme activities vary.
- Inactive: H46R & Gly85Arg
- Normal: C6S; E40G; L84F; Asp90Ala; G93A & G93D; D109Y
- Protein stability
- Unstable: L126X; G127X
- Normally stable: A89V; D90A
- Correlation between ratio of mutant:normal SOD1 in RBC
53
- Lower ratio: Shorter disease duration
- No correlation between duration of disease and SOD mutant peptide ...
- Ability to scavenge superoxide
- Half-life
- Solubility
- Resistance to proteolytic digestion
- Propensity to aggregate spontaneously into sedimentable structures
- Relative affinity for Cu
- Stop mutations all in exons 4 & 5: Active site in exon 3 preserved
- Allelic disorders
- SOD1 deficiency
- Animal variant: Myelopathy
- SOD1 protein
- Abundant: 1% of cytosolic protein
- Structure: Homodimer
- Each subunit contains Cu++ & Zn++ in cave-like active site
- Subcellular location: Cytoplasmic & mitochondrial
- Function
- Detoxifies O2- to H2O2
- Prevents oxidative damage
- Associated enzymes preventing oxidative damage: Catalase; Glutathione peroxidase
- Zn++ maintains pH stability of dismutase reaction
- Misfolded SOD1: Binds to VDAC1 78
- Other SOD molecules
- SOD2: Mitochondrial; Manganese
- SOD3: Extracellular; Copper-Zinc
- Clinical: General features of hereditary SOD1 ALS
- Clinical vs Sporadic ALS
- Fewer: Upper motor neuron signs
- Onset with monomelic leg weakness: Suggests familial ALS
- Most mutations with variable clinical features among patients
- Homozygosity
- Often causes more severe phenotype than heterozygous state: Asn86Ser; Asp90Ala
- Patients often have mainly lower motor neuron signs
- Onset
- Mean age
- General: 46 years
- US: 50 years
- China: 44 to 46 years
- 10 years younger than sporadic ALS
- 3.5 years younger than non-SOD FALS
- 90% penetrance by age 70
- General: 46 years
- Juvenile (< 25 years) onset: Few patients
- SOD1 Mutations: de novo; G16S, P66S, H80A
- Onset age: 18 to 24 years
- Course: Rapid progression
- No anticipation
- Preparetic phase: Some with myalgias, leg cramps & painful paresthesias
- Weakness: Asymmetric limb
- Mean age
- Duration
- Mean for all FALS = 3.4 to 5.6 yrs
- Similar for SOD & non-SOD FALS
- Survival
- Male > Female
- 5 years: 31% to 55%
- Varies strongly with specific mutations
- Lower motor neuron signs
- Common
- Especially predominant in rapidly progressive cases
- Upper motor neuron signs
- May be more common with slowly progressive course
- Reported with Asp76Tyr & Homozygous Asp90Ala mutations
- Dominant upper motor neuron signs: Not reported
- Bulbar onset
- Unusual
- Later onset age
- Reported with some mutations
- Cramps: G12R; G37R; D90A; E100G
- Other Neural features
- Sensory neuropathy: A89V; D90A
- Extra-motor system symptoms: ? More frequent
- Dementia: Rare; A4T; G41S; I113T; L144F; ? More common in non-SOD group
- Autonomic failure: D90A; Val118Leu; 1 patient 3
- Vocal cord dysfunction: A4V; I113F; Gly147Ser; I149T
- Supranuclear EOM paresis: May occur late in disease course; H80R; I104F
- Bladder dysfunction: G41S; Asp90Ala (Homozygous)
- Treatment
- SOD1 Antisense oligonucleotide 184
- Clinical vs Sporadic ALS
- Population statistics
| ALS syndromes: Correlations with some SOD1 mutations | |
|---|---|
|
Specific SOD1 mutations Exon 1; Ala4Val Most common mutation Rapid onset & progression (1.0 yrs) Frequently only lower motor neuron signs Exon 2; His46Arg @ Cu binding site of SOD Onset: Late; Legs Bulbar unusual Slow progression (17 yrs) Exon 2; 6 bp deletion(ΔG27/P28) 65 Mutation reduces transcription Low levels of mutant SOD1 protein Philipino founder Low penetrance Disease duration: 4.3 years Exon 4; Leu84Val Lower motor neuron only Rapid progression (1.5 yrs) ?Earlier onset in males Exon 4; Asp90Ala Onset: 20 to 94 yrs; Legs; Preparetic phase Leg cramps; Myalgia; Painful paresthesia Bladder dysfunction Progression: Slow; Legs ® Arms Inheritance Recessive: Finnish (2.5% carriers) Dominant: Clinically variable Incomplete penetrance Exon 4; Ile104Phe Variable intrafamilial clinical features Age of Onset: 6 yrs - asymptomatic Course: 2 to 14 yrs until bulbar signs Limb onset: arms or legs Exon 4; Ile113Thr Reported in Sporadic ALS patients Relatively common; Low penetrance Late Onset: Mean 59 years Course: Variable; 2 to 20 years Exon 5; Codon 126 2 base pair deletion Rapid Progression |
ALS clinical features Lower motor neuron predominant A4V; G72C; Leu84Val; Gly93Cys; E100K; D101N; S134N Slow progression C6S; Asp11Tyr; G12R; V31A; Gly37Arg (18 yrs); Gly41Asp (11 yrs); F45C; H46R131; D76V; Gly93Cys (13 yrs); Gly93Ser; Leu144Ser; Leu144Phe (9 yrs) Rapid progression Ala4Thr (1.5 yrs); A4V; C6G; C6F V7E; L8Q; G10V; G41S; H43R; H48Q Asn86Ser Homozygous (5 mo); D101>G,H,N,Y; Leu106Val (1.2 yrs); I112T; I113F; Arg115; D125H; 126 2bp del; S134N; Gly147Ser; Val148Gly (2 yrs); V148G Late onset His46Arg; Gly85Arg (55 yrs); D90A; I113T; Ala140Gly; Leu144Phe Early onset Gly37Arg; Leu38Val; A89V; L104F (6 yrs); ?Leu106Val More common in females Gly41Asp Bulbar onset Cys6Gly; L8Q; His48Gln; Asp76Tyr; Asp90Ala (Homozygous); D101Y; I112M; T116R; Cys146Arg; Gly147Ser; Val148Ile; I149T; Ile151Thr Onset in legs G10V; H46R; L84F; D90A; Gly93Cys; Gly93Ser SOD Mutations in "sporadic" ALS Most common: Asp90Ala; Ile113Thr Other: Asp11Tyr; V14G; G16S; E21K; G72S; D101N; V118InsAAAAC; E133delGAA Incomplete penetrance |
- Genetic variants: Other features
- Ala4Val
147
- Epidemiology
- Common among North American SOD1 ALS patients: 36% to 50%
- Unusual in China
- SOD1 protein: Folding of mutated SOD1 protein less stable than normal 30
- Clinical
- Onset age: Mean 50 years
- Course
- Rapidly progressive
- Mean survival: 1.4 years; 50% of other SOD1 mutations
- Lower motor neuron predominant
- Epidemiology
- Phe20Cys
60
- Onset
- Legs
- Weakness or Fasciculations
- Later in Females
- Course: Mean 1.9 years
- Bulbar signs present late
- Onset
- Gly85Arg (Transgenic mouse pathology)
- Rapid clinical course; Normal SOD1 activity
- Astrocytic inclusions: Contain SOD1 & ubiquitin; Increased with disease progression
- Aggregates in motor neurons: Contain SOD1
- Reduced Glial glutamate transporter
with disease progression
- ? 1° disease in astrocytes
- Asp90Ala
- Finnish population
- Other geographic locales
- Dominant inheritance
- Progression in heterozygotes: Typical ALS course
- SOD1 activity: Normal
- Rat SOD models
18
- G93A high expression
- Onset: 110 days
- Rapid disease progression (8 days)
- Motor neuron degeneration
- Vacuolar pathology
- H46R-4 high expression
- Onset: 140 days
- Slower progression (24 days)
- Motor neuron pathology
- Protein deposition & aggregation
- G93A high expression
- Other transgenic mice
- Mitochondrial vacuoles, or
- Neurofilament accumulation
- Mitochondrial vacuoles, or
- Ala4Val
147
- SOD1 MND Laboratory
- CSF protein: Often high, atypical for other ALS
- Pathology of SOD1-ALS: May vary with different mutations
- Common features: Some different from sporadic ALS
- Cell loss: Corticospinal tract neurons & Anterior horn
- Relatively normal corticospinal tract & brainstem motor nuclei: Gly93Cys
- Posterior column changes
- Especially fasciculus cuneatus (Middle root zone) at cervical level
- Mutations: A4V; A4T; Gly93Cys; Gly93Ser; I113T; Not H48Q
- Spinocerebellar tract pallor
- Mouse models
- Activation of non-neuronal cell types: Astroglia & Microglia
- Sequential activation of Caspase-1 and -3
- Cell loss: Corticospinal tract neurons & Anterior horn
- Hyaline conglomerate inclusions (HCI)
27
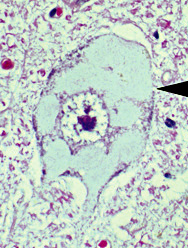
Hyaline conglomerate
inclusion- Cells: Surviving motor neurons
- Subcellular location: Intracytoplasmic
- Contain SOD, neurofilaments, peripherin ± ubiquitin
- No correlation between frequency of aggregates & cell death
- Mutations: Ala4Val, A4T, H48Q, D101N, I112T, I113T, 126 bp deletion
- Have specificity for SOD1 FALS mutations
- E100G: ALS-dementia type
- Dentate granule inclusions
- Frontal lobe gliosis
- Muscle
- Common features: Some different from sporadic ALS
- Animal variant: Canine Degenerative Myelopathy, Recessive SOD1
92
- Dog breeds (115): Pembroke Welsh corgi, Boxer, Rhodesian ridgeback, German Shepherd, Markiesje & Chesapeake Bay retriever
- SOD1 mutations
- Homozygous missense: E40K
- Most same mutation; Also Thr18Ser
- Wire Fox Terrier: High frequency of E40K allele; Low frequency of disease
- Clinical
- Onset age: ≥ 8 years
- Spastic paraparesis
- Gait ataxia
- Flaccid tetraparesis: Late
- Dyskinesia
- Course: Progressive over 2 to 3 years
- Pathology: Spinal cord
- Lateral white matter: Myelin & axon loss
- Neuronal cytoplasmic inclusions: Contain SOD1
- No motor neuron cell body loss
- SOD1 variant: SOD1 deficiency
155
- Nosology: Spastic Tetraplegia & Axial Hypotonia, Progressive (STAHP)
- Epidemiology: 1 Afghan family
- Genetics
- Mutation: Homozygous; Truncating; c.335dupG
- Inheritance: Recessive
- Heterozygous carriers: No phenotype
- Clinical
- Onset age: 9 months
- Developmental delay: Cognition imp[aired
- Dysmorphism: Low set ears; Overlapping toes
- Spasticity: Tetraparesis; Legs & Arms; Tendon reflexes brisk
- Hyperekplexia
- Laboratory
- Brain MRI: Cerebellar atrophy
- Blood manganese: Low
- SOD1 activity: Undetectable
- Muscle
- Fiber sizes: Varied
- α-dystroglycan: Focally reduced
- Muscle ultrasound & EMG: Fasciculations
- NCV: Normal
● Ubiquilin 2 (UBQLN2; PLIC2; CHAP1)
- Epidemiology
- Frequency: 5 families; ~1% of ALS
- Male vs Female
- Progression: Similar disease duration
- Onset age: Males younger; 34 vs 47 years
- Genetics
- Mutation locationss
- Missense in Proline: Pro497His, Pro497Ser, P506T, P509S, P525S
- In or near PXX (Proline/Glycine repeats) domain
- Sporadic patients: Outside PXX domain
- Gene subjected to X-inactivation
- Penetrance: 90% by age 71
- Variant syndrome: Spastic paraparesis
- Female carriers: Usually asymptomatic
- Mutation locationss
- UBQLN2 protein
- Ubiquitin-like protein family: Bind to subunits of proteasome
- Regulates: Protein degradation pathways
- Ubiquitin-proteasome system (UPS)
- Endoplasmic reticulum�associated protein degradation pathway: Interacts with UBXD8 & HERPUD1
- Macroautophagy
- Interacts with: TDP-43; HSP70 (HSPA1A)
- Mutations: May impair autophagy
- Clinical
- General phenotypes: ALS, ALS + FTD, Juvenile-onset ALS, Madras
- Onset
- Age: 10 to 71 years; Older in females
- Bulbar dysfunction
- Dementia: Some patients
- Upper motor neuron
- Spasticity: Legs > Arms; Gait disorder
- Tendon reflexes: Brisk
- Bulbar & Pseudobulbar dysfunction
- Prominent
- Dysarthria
- Dysphagia
- Lower motor neuron: Often not prominent
- Weakness: Plantar foot flexion; Hands in some patients
- Fasciculations: Some patients
- Wasting: Occasional
- Dementia
- Some patients
- Fronto-Temporal, then Global
- May precede motor signs
- Sensory: Normal
- Progression
- Slow
- Death after 2 to 17 years
- Laboratory
- CNS pathology
- Motor neurons: Skein-like inclusions
- Contain Ubiquilin-2, Ubiquitin, p62, TDP43, FUS, Optineurin
- No SOD1
- Spinal cord
- Axon loss: Corticospinal tract
- Anterior horn: Loss of anterior horn cells; Astrocytosis
- Hippocampus: Ubiquillin-2 pathology in ALS/FTD with & without UBQLN2 mutations
- Motor neurons: Skein-like inclusions
- CNS pathology
- Variant phenotype: Madras motor neuron syndrome
● Fusion, derived from 12-16 translocation, malignant liposarcoma (FUS; FUS/TLS)
- Epidemiology
- Multiple families: More common in Asia
- Familial ALS: Frequency 3% to 5%
- Common in: Early-onset (Juvenile) ALS, Familial or Sporadic
- Sporadic ALS
- Frequency
- General ALS: 0.3% to 0.7%
- Juvenile onset
- 7% to 50%
- More in Japan & China
- Most common gene with fALS onset < 25 years
- Mutations: Exon 6; G507D, R518G, R521C, R521H
- Frequency
- Genetics
- Mutations
- Missense, In-frame deletion or insertion, or Frameshift
- Hot spots: Exon 15; Arg521; Pro525
- Other mutations: > 30; In Exons 3, 5, 6 & 14
- Recessive mutation: H517Q
- R514S & R521C: Associated with Symmetric, Proximal arm, neck & axial weakness
- Pro525Leu & Pro525Arg
144
- Onset age: Very young (<30 years)
- Bulbar presentation
- Disease duration: Short, Rapid progression
- Cytoplasmic inclusions: Stress granule�like
- Pro525Leu: German patients
- Arg521: Less bulbar involvement
- Y526C: Young onset; Rapid progression
- Basophilic inclusions: P525L; c.1554-1557delACAG
- C-terminal nuclear localization truncation mutations: More aggressive disease
- de novo mutations: Described in early onset (11 to 34 years) ALS syndromes
- Incomplete penetrance: Common
- Allelic disorders
- Essential tremor 4
96
- Epidemiology: Rare families
- Mutations: Gln290X; Arg216Cys; Met392Ile; Pro431Leu
- FUS mutant proteins: Degraded by nonsense-mediated-decay pathway
- No symptoms of ALS
- Frontotemporal dementia
- ALS/Parkinson disease
- Juvenile-onset ALS (< 25 years)
- fALS, Recessive
- ALS6, Dominant
- ALS3: ALS family with gene originally localized to 18q21
17
- Essential tremor 4
- Mutations
- FUS protein
177
- Cellular localization
- Normal: Nuclear; Synapses (Pre- & Post-synaptic)
- Disease
- Cytoplasm accumulation
- Present in: Anterior horn cell aggregates in sporadic & hereditary ALS except SOD1
- Protein groups
- Functions
- DNA & RNA metabolism
- DNA-/RNA-binding
- DNA repair
- RNA
- Interacting domains: Located in the mid-region of protein
- Processing: Regulation of splicing & transcription of synaptic mRNAs
- RNA binding: Introns, evolutionary conserved
- Interactions
- Syntaphilin (SNPH)
: Mitochondrial tethering protein
- Mitochondrial proteins
- GluA1 (GRIA1)
- U7 snRNA/snRNP: Regulates expression of histone genes
- snRNP70
- Gene spicing variants
- Alternative 3'UTR selection
- Associated with ALS subtype: Increased oxidative & proteotoxic stress
- Gene spicing variants
- U1 snRNA & U1 snRNP
- Syntaphilin (SNPH)
- Implicated in
- Tumorigenesis
- RNA metabolism
- Stress granule formation
- FUS aggregates: 10% of FTD
- FUS deficient neurons: Decreased spine arborization with abnormal morphology
- Hippocampal neuronal slice cultures: FUS is in RNA granules that are transported to dendritic spines
- Local RNA translation in response to metabotropic glutamate receptor (mGluR5) stimulation
- DNA & RNA metabolism
- Mutations: Often have abnormal cytoplasm localization
- Knockout mice: Perinatal mortality; Male sterility; Chromosomal instability; Radiation sensitivity
- Cellular localization
- Onset
- Age
- Mean 37 to 44 years; Range Teens to > 70 years
- Variable penetrance at 70 years: 39% to 83%
- Earlier & shorter survival than SOD1 & TDP43 FALS
- Upper cervical & neck
- Limbs or Bulbar: Bulbar more frequent than SOD1 FALS
- Age
- General phenotypes: Predominantly lower motor neuron syndromes
- Axial & arm weakness: Dropped head; Mid to late adult onset
- Benign: Slow progression; Late onset
- Juvenile: Bulbar onset; Cognitive Δ
- Genotype-Phenotype correlations: Mutation locations
- Last 2 amino acids in NLS domain: Aggressive phenotype; Bulbar onset; Cognitive Δ
- Missense at 510 to 521 (K510R, H517Y, Q519E, R521H): Mild; Slow progression; Flail leg
- Missense in C-terminal (Common): Proximal arm; LMN phenotype
- P525L & Y526L: Juvenile
- Lower motor neuron signs
- Common
- Weakness: Distribution
- Limbs
- Bulbar: May be present at onset
- Early: Proximal or bulbar in some patients; Upper or Lower extremities
- May be symmetric or asymmetric
- Posterior neck
- Upper motor neuron signs: Common
- Other CNS
- Cognition: Usually normal; Defecits in few patients
- Extrapyramidal: Parkinsonism in few patients
- Progression
- Variable among families: Range 0.5 to 20 years; Mean 3.4 years
- Slower progression: Later onset age
- More rapid than SOD1 ALS: 89% die in < 4 years
- Neoplasm association: None
- Loss of motor neurons: Anterior horn of spinal cord & hypoglossal nucleus
- Myelin pallor in anterior corticospinal tracts
- Macrophages surrounding shrunken Betz cells in motor cortex (neuronophagia)
- Increased lipofuscin staining in neurons
- Epidemiology: Cape Verdean family
- FUS mutation: Homozygous H517Q
- Clinical
- Onset: Proximal upper extremities
- Spread: To lower extremities; Not bulbar region
- Progression: Slow; Up to 14 years
- Family history: None
- FUS mutations
- C-terminal region
- P525L (c.1554-1557delACAG); Y526C
- Clinical
- Laboratory
- Pathology
- CNS: Loss of axons in motor roots & corticospinal tracts
- Basophilic inclusions
- In motor & non-motor neurons
- Stain for p62, PABP & FUS
- Tubulofilamentous structures
- Associated with electron-dense granules
- ? Origin from endoplasmic reticulum
- EMG: Diffuse denervation
- Pathology
● Chromosome 20p13; Dominant
- Epidemiology: 1 family
- Genetics
- Linkage security: Probable
- Clinical
- Onset age: Mean = 57 years
- Clinical: ALS = El Escorial criteria
- Course: Progressive; Mean = 3 years
● Vesicle-associated membrane protein B (VAPB)
- Epidemiology
- 7 Brazilian families
- Rare in other populations
- Genetics
- Same missense mutation in all families: Pro56Ser
- Allelic with: Proximal SMA, Adult onset
- VAPB protein
- Location: Associated with ER & Golgi membranes
- Lipid-droplet-ER contact
- Regulates membrane delivery into dendrites.
- Mutant protein: Intracellular aggregates not associated with membranes
- Clinical: 3 variant syndromes
- Onset
- Age: 25 to 55 years
- Cramps
- Fasciculations
- Weakness: Few patients
- Heterogeneity: Inter- & Intra-familial
- Atypical ALS
- Onset: 25 to 44 years
- Bulbar signs: Dysphagia (73%)
- Lower motor neuron
- Weakness: Arms & Legs; Trunk & Tongue in some patients
- Cramps: Long-standing; Painful; Easily precipitated
- Fasciculations
- Upper motor neuron: Few patients
- Tendon reflexes: Reduced
- Postural tremor
- Sensory loss: Few patients
- Course
- Slowly progressive
- 1 patient with respiratory failure
- ALS, Rapidly progressive
- Survival: < 5 years
- Lower motor neuron signs
- Pyramidal signs
- Occurs in same kindreds as atypical ALS syndrome
- Spinal muscular atrophy, Late onset
- Onset 35 to 55 years
- Weakness: Predominantly proximal
- Fasciculations
- Cramps
- Tendon reflexes: Reduced or Absent
- No bulbar or pyramidal involvement
- Onset
- Laboratory
- Serum CK: Normal or Mildly elevated
- EMG: Denervation, proximal, distal & tongue; Giant motor unit potentials
- NCV: Normal motor & Sensory
- Muscle biopsy: Neurogenic
- Groups of large & small angulated fibres
- Fiber type grouping
● Angiogenin (ANG)
- Epidemiology
- Most identified in Irish & Scottish populations
- Uncommon in US
- Mutations found in some patients with no family history of ALS
- Genetics
- Missense mutations: Q12L; K17I; K17E; R31K; C39W; K40I; I46V; V113I
- Similar mutations occur in some Parkinson patients
- Some missense mutations found in healthy controls: I46V; K17I
- Angiogenin protein
- Mediator of blood vessel formation
- Promotes endothelial invasiveness needed for blood vessel formation
- Induces vascularization of normal and malignant tissues
- Required for VEGF activity
- Ribonuclease family: Abolishes protein synthesis by hydrolyzing cellular tRNAs
- Induced by hypoxia
- Transported to nucleus after uptake by endothelial cells
- Present in wide spectrum of cells: Especially liver
- Cell localization
- Contains nuclear localization signal
- Secreted protein
- Mutation effects: Loss of function
- Mediator of blood vessel formation
- Clinical
- Onset
- Age range: 27 to 76; Similar to ALS or PD without mutation
- Bulbar onset: 50%
- Spinal onset: 50%
- Pattern: "Typical ALS"; No specific phenotype
- Upper motor neuron
- Lower motor neuron
- Course: 0.8 to 10 year survival
- Parkinson disease
- Often occurs with no ALS
- Some ALS syndrome patients have Parkinson signs
- Onset
● Senataxin (SETX)
- Epidemiology: England; Southern Maryland
- Genetics
44
- Mutations
- Missense
- Locations: T3I, Glu385Lys, Leu389Ser, R2136H
- Differ from stop or truncation mutations in senataxin in AOA2
- Allelic disorders
- Distal HMN with upper motor neuron signs
- AOA2 (Recessive)
- Similar locus to
- Mutations
- Senataxin protein
- Clinical
- Onset age: 2nd decade; Mean = < 6 to 21 years
- Early: Gait disorder
- Weakness
- Distal: Later also proximal
- Hands & Feet
- Usually symmetric
- Bulbar disorders: Infrequent, Dysphagia
- Muscle wasting: Distal
- Upper motor neuron signs
- Hyperreflexia 86%
- Upgoing toes 17%
- Sensory: Normal or Mild changes in some (30%) older patients
- CNS: Ataxia in arms in up to 50%
- Severity: Variable degrees
- Course
- Slowly progressive over decades
- 5th & 6th decades: Wheelchair; Loss of hand function
- Milder phenotype: Mild gait disorder
- Laboratory
- Electrophysiology: Motor neuropathy, Chronic
- EMG: Denervation, Distal > Proximal, Chronic
- NCV
- CMAP amplitude: Reduced
- Distal latencies: Long
- Temporal dispersion: Some patients
- Conduction block: some patients
- NCV: Normal
- Sensory: Normal
- Pathology
- Reduced number of Anterior horn cells, especially lumbar
- Sensory pathways: Posterior column fiber loss; Loss of DRG neurons
- Axon swellings: Roots, Spinal gray, Dentate, Cranial nerves 3 & 4
- Spinal cord atrophy
- Muscle MRI: Fat increased in legs
- Serum CK: Normal or Mildly high
- Colon polyps (25%)
- Knock in mice: CD8 cell clonal increase
- Electrophysiology: Motor neuropathy, Chronic
● C9orf72
- Epidemiology
- Frequency
- High frequency
- Europe & America
- 60% of hereditary ALS-FTD
- 27% to 50% of familial ALS patients
- Finnish ALS patients: 50% of familial; 21% of sporadic
- Sweden
- Jewish
145
- Ashkenazi
- Present in up to 80% of fALS
- More common in bulbar onset sALS
- Have nucleotide repeat expansion on common, unstable risk haplotype
- North African Jews
- Other Jewish fALS Association: OPTN
- Ashkenazi
- C9orf72 expansion in UK population: 1 in 700
- Europe & America
- Low frequency: Germany
- Rare: Native Americans; Iran; Asia; Pacific Islands
- Sporadic (Simplex) ALS patient frequency
- ALS
- General: 0% to 21%
- High in Finland
- China: 0.8%
- FTLD: Sporadic 7% to 10%
- ALS
- FTLD familial: 19%
- Lifetime risk of C9orf72-associated FTLD or ALS: 1 in 2,000
- High frequency
- Sex: Male = Female
- Transmission from females: 1.4x higher
- Frequency
- Genetics
- Mutation: GGGGCC (G4C2) hexanucleotide repeat expansion
- Location
- 1st intron & promoter
- Non-coding region between non-coding exons 1a & 1b
- Structurally diverse
- Hexanucleotide repeat numbers
- Controls
- Range
153
- Control populations: 2 to 45; Rarely as high as 70
- Unaffected individuals
- General: All < 25
- Size range: 2 to 25
- Most common repeat sizes: 2 or 3
- Other peak lengths: 5 & 8
- ≤ 10: 94%
- Intermediate lengths: 7 to 45
- Finland: Repeat lengths > 20 more common
- Repeat type: Uninterrupted
- Linkage disequilibrium between repeat length & neighboring SNP rs3849942
- Intergenerational repeat change
- Rate: 0.29%
- Intergenerational changes (Sequence instability)
- Usually occur from starting repeat length ≥ 8
- No expansion into disease range
- All occurred on rs3849942A haplotype
- Range
153
- Patients
- Range, General: 250 to 4,400
- Symptoms reported with 20 to 22 repeats
- Somatic instability
- Present
- Different expansion size in varying brain & tissue areas
- No recent shared ancestry
- 70 repeat expansions
- Increased C9orf72 expression
- On typical 200-kb risk haplotype
- May increase to pathogenic size range (~1,750 repeats) across generation
- Controls
- Location
- Associated genetics
- Associated with SNPs: rs2477521; rs3849942 allele A
- Finnish founder risk haplotype: All cases with GGGGCC repeat expansion
- Hypermethylation of CpG island near & 5' of G4C2 repeat
103
- Associated & segregates with G4C2 expansion
- Higher degree of methylation: Shorter disease duration
- No methylation for normal or intermediate alleles (up to 43 repeats)
- Penetrance
142
- Younger ages: Low; Rare < 35 years
- 58 years: 50% penetrance
- Older ages
- 83 years: 75% to 99% penetrance
- Females 2 years older than Males
- Female + Bulbar onset: 3 years older
- ALS: Mildly earlier onset age than FTD
- Spinal onset: 2 years earlier than Bulbar
- Genotype-Phenotype correlations & modifiers
- c9orf72 expansion size
- Greater size in blood
- Correlates with: Older age at clinical onset
- No relation to: Clinical syndrome diagnosis
- Size of repeat in brain: Similar in most c9orf72 mutation patients
- Sporadic ALS
189
- c9orf72 repeat length ≤ 2: Longer survival
- c9orf72 repeat length longer in normal range: Shorter survival with bulbar onset
- Greater size in blood
- Frontotemporal lobar degeneration with TDP-43 pathology (FTLD-TDP)
- c9orf72 mutations are most common cause
- TMEM106B
homozygous minor rs3173615 allele (GG; p.T185S)
108
- Protects (OR 0.26) c9orf72 mutation carriers from developing FTD
- No protection from MND
- Also protective (OR 0.12) of FTLD-TDP
caused by GRN
mutations
- TMEM106B A rs1990622 allele: Later age onset
- ATXN2 intermediary polyQ expansions
116
- Associated with development of clinical signs of both FTD & ALS
- Predispose to: Expression of motor neuron disease within phenotype
- Frequency in c9orf72 carriers: 3%
- c9orf72 expansion size
- c9orf72 expansions can also occur with clinical diagnoses of
- Alzheimer disease (1.2%)
- Sporadic Creutzfeldt-Jakob disease (0.2%)
- Huntington disease-like syndrome (1.7%)
- Nonspecific neurodegenerative disease syndromes (2%)
- Ataxia
- Corticobasal syndromes
- Psychiatric disorders: Suicide may be common 163
- Controls (0.15%)
- r(GGGGCC)n RNA repeats: Possible disease mechanisms:
196
- C9orf72 protein haploinsufficiency (Down regulation)
- Bidirectionally transcribed
- Sense (G4C2) & Antisense (C4G2)
- Into RNA foci: Nuclear
- RNA: Binds to & disrupt RNA splicing, transport & translation
- Non-canonical translation
- Dipeptide repeats (poly-GA, -GP, -GR, -PA, -PR)
- Inclusions or toxicity
- Toxicity
- Directly bind & sequestrate other proteins
- Interfere with: rRNA synthesis, Ribosome biogenesis, translation & nucleocytoplasmic transport
- Mutation: GGGGCC (G4C2) hexanucleotide repeat expansion
- C9orf72 expression
107
- Subcellular: Probably nuclear
- Cellular: Neurons > > Astrocytes or Microglia
- Gray matter > White matter
- Functions through binding to
- Smith-Magenis chromosome regions 8 (SMCR8)
- WD repeat-containing protein (WDR41)
- Regulates autophagy
- Mutation
- Reduced expression of isoform a
- Associated with formation of nuclear RNA foci
- Clinical
157
- Onset
- Age
- General
- Range: 34 to 84 years
- Mean: 6th decade; 55 to 59 years
- Median: 58 years
- Similar for: ALS & FTD presentations
- Same, or earlier, mean age as: Sporadic ALS
- Young onset (< 45 years): Less than singleton ALS
- Anticipation: 2 to 3 years younger onset per generation 137
- Earlier onset: Sporadic cases 163
- Later onset: More G4C2 repeats
- General
- Clinical
- Weakness
- Limb
- Frequency: 50% to 67%
- Legs: More common
- Arms: Less common
- Bulbar
- Frequency 33% to 43%
- More common than sporadic ALS
- Clustered in some families
- Motor features before Dementia (30%)
- Limb
- Cognitive
- Dementia (FTD) before Motor (32%)
- Psychosis
- Paranoid, deluded or irrational thinking
- Weakness
- Age
- Motor neuron disease
- Occurs in patients without FTD (> 50%)
- Upper motor neuron (95%)
- Tendon reflexes: Brisk (95%)
- Spasticity (46%)
- Plantar response: Upgoing in 20%
- Bulbar dysfunction
- Lower motor neuron
- Weakness
- Fasciculations
- Course
- Survival: Median 30 to 37 months
- Monthly change: -1.4% (sVC) to -1.8% (ALS-FRS)
- vs sporadic ALS: Fewer slowly progressive patients
- No corrrelationi with repeat size
- Clinical variants
109
- No dementia: 50%
- Primarily upper or lower motor neuron involvement: Rare
- Spastic paraparesis: Rare
- Dementia (FTD)
- Frontotemporal lobar degeneration (FTLD)
- Frequency
- Patients: 30% to 35%
- Families: 50%
- Type
- Behavioral (64%)
- Progressive nonfluent aphasia (27%)
- Semantic dementia (9%)
- Psychiatric symptoms: 50%
- Apathetic
- Socially isolated
- Delusion
- Working memory: Impairment
- Less eating dysregulation than FTD non-carriers
- Cortical
- Cognitively impaired
- Limb apraxia
- Cortical sensory loss
- Aphasia: Non-fluent
- May occur in some patients without motor neuron disease
- Extrapyramidal
- Increased tone, Some patients
- Huntington disease phenotype (2%)
- Onset age: Younger than other c9orf72 syndromes; May be pediatric
- Signs: Dystonia, Chorea, Myoclonus, Tremor, Rigidity
- Epilepsy: Myoclonic
146
- Epidemiology: 1 family
- Genetics
- Mutation: c9orf72 repeat expansion
- Other family members with psychiatric or neurodegenerative disorders
- Onset age: 15 years
- Disease duration
- ALS
- Mean 3.6 years; Range 1�6 years
- Long survival: Less frequent than singleton ALS
- FTD: Mean 5 years; Range 1�12 years
- ALS
- Onset
- Laboratory
- EMG: Denervation
- Muscle: Neurogenic atrophy
- MRI
- Atrophy
- Frontotemporal: Bilateral
- Thalamus
- FTD patients: May have presymptomatic cortical thinning
- Fusiform, Thalamic, Supramarginal, Broca & Orbitofrontal regions: Involved in c9orf72, but not sporadic, ALS
- Hypoperfusion: Frontal & Temporal; bilateral
- White matter: Corpus callosum, Superior motor tracts, Corticospinal & Cerebellar pathways
- Atrophy
- CNS pathology
- Brain atrophy: Especially frontal
- Corticospinal tracts: Reduced myelin staining
- LMN: Loss of from brainstem & spinal cord
- Inclusions
- Staining
- Ubiquitin, p62 or TDP-43: Vary in different brain locations
- Ubiquitinated in anterior horn cells, Bunina bodies or Hirano bodies
- TDP-43-positive neuronal cytoplasmic: Cortex & Spinal cord
- Not tau positive
- Locations
- Neocortex
- Hippocampal dentate granule cells
- Cerebellar granule layer
- Intranuclear inclusions
- Transcribed GGGGCC repeat: Forms nuclear RNA foci in cortex & spinal cord
- Proteins: None described
- Staining
● Chromatin-modifying protein (CHMP2B)
- Epidemiology
- Sporadic ALS
- ALS: 1%
- Lower motor neuron predominant ALS (PMA): 10%
- Genetics
- Mutations
- Missense
- Ile29Val; Thr104Asn; Gln206His
- Other mutations cause: Fronto-Temporal Dementia
- Mutations
- Protein
- Clinical
- Onset
- Age: 7th & 8th decade
- Bulbar dysfunction
- Lower motor neuron syndrome: (Ile29Val; Thr104Asn; Gln206His)
- Clinical
- Weakness
- Bulbar (Tongue)
- Dysarthria
- Weakness
- Wasting
- Hands or Legs
- Respiratory
- Bulbar (Tongue)
- Fasciculations: Tongue & Limbs
- Tendon reflexes: Reduced or Normal
- No UMN signs
- No FTD
- Progression: Respiratory failure over 15 months
- Weakness
- EMG: Diffuse denervation
- Clinical
- I29V mutation: Fronto-temporal dementia syndrome (FTD-3)
- May be benign variation or pathogenic mutation
- Clinical
- Onset: Dementia
- Spastic dysarthria & dysphagia
- Weakness: 1 arm & both legs
- Tendon reflexes: Brisk
- Onset
- Pathology
123
- Motor neurons
- Loss
- Ubiquitylated inclusions
- p62 antibodies (sequestosome 1): Oligodendroglial inclusions in motor cortex
- Neuronal Pathology: Frontal cortex
- Lysosomal: Autofluorescent aggregates
- Motor neurons
● Oncogene DJ-1 (PARK7; DJ1)
- Epidemiology: Southern Italian & Turkish families
- Genetics
- Mutations: Q45X; E163K & g.168_185dup
- Allelic disorder: Parkinson disease 7, Autosomal recessive, Early-onset
- Clinical
- Onset age: 3rd & 4th decades
- Parkinsonism
- Early onset
- Bradykinesia
- Tremor
- Asymmetric
- Levodopa responsive
- Some families with only PD
- Motor system
- Weakness: Arms & Legs
- Hand atrophy
- Tendon reflexes: Brisk
- Plantar response: Extensor
- Cognitive impairment
- Laboratory
- EMG: Fibrillations; Fasciculations
- NCV: SNAPs normal
- CSF: Normal
● Autosomal Dominant
- Epidemiology: Japanese family
- Clinical
- Onset
- Bulbar dysfunction
- Age: 5th & 6th decade
- Cranial nerves: Atrophy of facial muscles & tongue; Dysarthria
- Weakness: Arms ± Legs; Respiratory
- Tendon reflexes increased
- Normal: Sensation; Intellect; Bladder
- Course: Progressive over 10 years
- Onset
- Pathology
- Reduced numbers of Upper & Lower motor neurons
- Bunina bodies: In lower motor neurons
- Normal: posterior columns
● Neurofilament heavy chain (NEFH)
- Genetics
- Mutation type: Inframe deletion or insertion
- Mutation location
- Large NFH side arm domain
- Hypervariable C-terminus
- Amino acids Lys-Ser-Pro (KSP)
- KSP sequences in normal neurofilament heavy chain
- Repeated > 40 times
- Located in hypervariable region
- Commonly phosphorylated
- Sites for proline-directed serine/threonine protein kinases
- Located in sidearm projection that cross links neurofilaments
- Mouse mutants
- Over expression of neurofilaments: Associated with motor neuron disease
- Neurofilament subunit knock-out: Reduced axon caliber
- Allelic disorder: CMT, Axonal
- Neurofilaments
- Clinical (Genetic associations)
- Sporadic ALS: Mutations in 1% of patients
- Role in disease: ? Risk factor rather than causative effect
- Reduced expression of NFL in sporadic ALS patients without SOD1 or NF mutations
- Tetranucleotide repeat: TTTA in intron
190
- Repeat range: 6 to 15
- 9 TTTA allele: sALS risk reduced; Spinal onset ALS
- 10 TTTA allele; 2.7 yr later onset vs othe sALS
- Hereditary ALS: Scandinavian pedigree
- ALS in 4 members of 2 generations
- Deletion in the NF-H tail: Nucleotides 1989-2030
- Clinical features: Variable
- Dementia
- Dysphagia
- Monomelic ALS
- Sporadic ALS: Mutations in 1% of patients
- NEFH variant syndrome: CMT 2CC, Axonal
129
- Epidemiology: 15 families
- Genetics
- Inheritance: Dominant
- Mutations: Frameshift (Stop) in 3' UTR or C-terminus of NEFH; 1 missense
- Mutation effects
- Translation of cryptic amyloidogenic elements (CAEs) encoded by 3' UTR
- NEFH protein with 40 additional amino acids
- Aggregation-prone NEFH molecule
- Clinical
- Onset age: 2 to 64 years
- Weakness
- Distal or Proximal
- Legs > Arms
- Triceps; Quadriceps
- Muscle wasting: Distal
- Sensory loss
- Varied degrees
- Panmodal
- Distal
- Legs > Arms
- Tendon reflexes: Reduced in legs
- Upper motor neuron: Some patients
- Course: Slow progression
- Laboratory
- Serum CK: Normal to 1,288
- Brain & Spinal cord MRI: Normal
- Muscle pathology
- Denervation: Grouped atrophy; Fiber type grouping
- Internal nuclei
- Rimmed vacuoles: Few fibers
- Mitochondrial oxidative enzymes: Reduced complexes I, II+III & IV
- Nerve pathology
- Axon loss
- NCV: Axon loss, Legs > Arms; CMAP & SNAP amplitudes reduced
- EMG: Chronic denervation, Distal & Proximal; Proximal myopathic changes
- CSF: Normal
- Muscle MRI: Sparing of gracilis & biceps, short head in thigh
● Tar DNA-binding protein (TDP-43; TARDBP)
- Epidemiology
- Families: American, Canadian, English, French, Chinese
- More common in Southern (Sardinia) than Northern European populations
- 33% of ALS in Sardinia (A382T mutation)
- Familial ALS: 3% to 6%
- Sporadic ALS patients: Frequency ~1 in 200; 8 patients described
- 18% with TDP43 mutations sporadic
- Genetics: Mutations
- > 60 identified
- Type: Missense
- Locations
- Often exon 6
- Contains glycine rich domain
- C-terminal
- Interacts with splicing proteins (hnRNPs)
- G287S; Gly290Ala; Ser292Asn; Gly294Ala; Gly298; Ala315Thr;
Glu331Lys; Met337Val; G348C; R361S; A382T; N390D; N390S
- Often exon 6
- C-terminal prion-like domain (PrLD)
- Modulate liquid condensation & aggregation properties of TDP-43
- Other mutation: D169G (Exon 4)
- Clinical Correlations
- Shortest survival: G298S
- Long survival & Late onset: A315T; M337V
- Frequent mutations: G348C; A382T
- Southern China: Gly298Ser
- Mutation effects: May increase sTDP-43 isoform
- Allelic disorders
- Frontotemporal lobar degeneration with TDP-43 inclusions, TARDBP-related
- Myopathy with Rimmed vacuoles
- Other
- Frontotemporal lobar degeneration with TDP-43 inclusions, TARDBP-related
- Expression
- Ubiquitous
- Short isoform (sTDP-43): More prominent in
- Motor neurons
- Cytoplasm
- Subcellular distribution
- Nucleus & Cytoplasm
- Reduced in nucleus in cells with TDP-43 cytoplasmic aggregates
- Properties
- Tendency to self associate & form aggregates
- Sequestered into polyglutamine inclusions
- Phosphorylated TDP-43: Present in neuronal aggregates in ALS & FTD
- Contains: Prion-like domain
- hnRNP protein: Similarity to hnRNPs
- Stress granule related proteins (hnRNP): ALS related
- Tendency to self associate & form aggregates
- Binds DNA & RNA
- Especially in nucleus
- Binds to: Uridine/guanine (UG)-rich motifs in RNA transcripts
- Regulates: Transcription & Splicing
- Lack of RNA binding: May predispose to relocation of TDP-43 to cytoplasm
- Other TDP-43 actions
- Repressor of cryptic exons during splicing
- May be involved in microRNA biogenesis, apoptosis & cell division
- Sporadic ALS: TDP-43 pathology
- Nucleus: TDP-43 reduced
- Cytoplasm: TDP-43 aggregates common
- TDP-43 pathology related to increased mtDNA copy number
- Mutations
- TDP-43 mutation locations: C-terminal in region involved in protein-protein interactions
- Mutation effects
- Loss of TDP-43 from nucleus
- Dysfunctional TDP-43: Inclusion of cryptic exons into mRNAs; RNA stability decreased
- Alternative polyadenylation: Abnormal
- General: Onset age earlier; Disease duration longer
- Onset
- Age
- 4th to 8th decade
- Younger than sporadic ALS
- Weakness
- Arms: 50%; More than SOD1 ALS
- Legs: 20%
- Bulbar: Common in Asians
- Age
- Weakness
- Arms before Legs: 60%
- Distal + Proximal
- Respiratory
- Bulbar: Few patients
- Upper motor neuron
- Tendon reflexes: Brisk
- Spasticity: Mild or None
- Cognition
- Usually normal
- FTD-ALS syndromes: Occasional patient
- FTD may be presenting syndrome
- Other family members may have pure ALS with normal cognition
- More involved in Sardinia
- Extrapyramidal: Parkinsonism in few patients
176
- Tremor, resting
- Rigidity
- Akinesia
- Course
- Slowly progressive (5 to 10 years) in most
- Rapid (1 to 2 years) in some families (Gly290Ala; Gly298Ser)
- Mean: Longer survival than sporadic ALS
- Nerve conduction testing: Normal
- EMG: Denervation, proximal & distal; Fasciculations
- CNS pathology
- Anterior horns of spinal cord: Neuronal loss, Gliosis, and Bunina bodies
- Corticospinal tracts: Pallor
- Betz cell loss in primary motor cortex
- TDP-43 inclusions
- Locations: Upper & lower motor neurons + elsewhere in CNS
- Often ubiquitinated
- Neuronal, Cytoplasmic: Also Nuclear depletion of TDP-43
- Epidemiology: 1 Sardinian patient
- Genetics: A382T missense mutation
- Incomplete penetrance
- Clinical
- Onset age: 49
- Amyotrophic lateral sclerosis
- Bulbar: Dysarthria & Dysphagia
- Spasticity: Arms & Legs
- Tendon reflexes: Increased
- Plantar reflex: Extensor
- Parkinsonism: Rigidity; Bradykinesia
- Dystonia
- Tics: Motor & Vocal; Childhood onset
- Dementia: Frontotemporal
- Laboratory
- EMG: Distal denervation in arms & legs
- NCV: Reduced CMAP amplitude
- Parents: Heterozygous; Normal or Dementia
- Epidemiology: 4 families
- Genetics
- Inheritance: Dominant
- Mutations: Frameshift (Trp385IlefsTer10) or Missense (Gly376Val); PrlD/LCD domain
- Protein produced by Frameshift mutation: C-terminal altered PrLD
- Gly376Asp variant: Produces ALS
- Clinical
- Onset age: Adult; 3rd to 7th decade
- Weakness
- Legs > Arms
- Proximal + Distal or Distal predominant
- Forearm: Anterior compartment (Hand & Finger extensors)
- Myalgias
- Exercise intolerance
- Paresthesias: Distal legs
- Course
- Slow progression; Wheelchair 45 to 60 years
- No ALS
- Non-penetrance in 8th decade described
- Laboratory
- Muscle
- Fiber sizes: Varied
- Sarcoplasmic inclusions: TDP-43 & p62 positive
- Endomysial connective tissue: Increased
- Ultrastructure: Autophagosomes
- Transcriptomes: Abnormally spliced sarcomeric genes (TTN & NEB); Increased expression of muscle regeneration genes
- EMG: Motor unit potentials usually small amplitude & polyphasic
- Muscle MRI: Atrophy; Fat replacement
- Serum CK: Mildly high
- Neuropathy: Axon loss in some patients
- Muscle
- ALS + Extrapyramidal symptoms
- ALS + Supranuclear palsy
- Supranuclear palsy
- FTD without motor neuron disease
● Peripherin (PRPH)
- Genetics
- Mutations: 4 identified
- Nucleotide insertion in intron 8
- 1-bp deletion in exon 1: Frameshift
- Missense: R133P; D141Y
- Allelic disorder: Axonal neuropathy
- Mutations: 4 identified
- Peripherin protein
- Intermediate filament (type III)
- Location: Peripheral nervous system; Lower levels in motor neurons
- Increased levels after neuronal injury
- Overexpression in mice: Motor neuron disease
- Mutations disrupt neurofilament assembly
- Associated with ubiquitinated inclusions
- Round & Lewy body-like inclusions
- Hyaline conglomerate inclusions
- Axonal spheroids: Occur in proximal axons of diseased motor neurons
- Clinical: 4 patients
- Onset age: 57 years
- Course: 5 years
- Onset: Asymmetric leg weakness
- ALS diagnosis
- PRPH variant disorder: Sensory-Motor neuropathy
154
- Epidemiology: Icelandic population
- Genetics
- Inheritance: Recessive
- Mutation: Splice-donor variant; c.996+1G>A; Loss-of-function
- Clinical
- Neuropathy (50%)
- Onset age: 20 years
- Sensory
- Pain & Vibration loss
- Subjective: Foot numbness
- Weakness: Toe extensors
- Neuropathy (50%)
- Laboratory
- Sural NCV
- SNAP Amplitude: Smsll
- Conduction velocity: Normal
- Heterozygotes: Partial reduction in sural SNAP amplitude
- Sural NCV
● FIG4
- Epidemiology
- 10 patients of European ancestry
- Frequency: 1% to 2% of ALS patients
- ALS
- Mutations
- Heterozygous
- Missense or Termination
- No patient with Ile41Thr mutation common in CMT 4J
- Allelic disorders
- Mutations
- FIG4 protein
- Clinical patterns
- Onset age: Mean 56 years; Range 29 to 77 years
- ALS or PLS
- Onset site: Bulbar, Legs or Arms
- Brisk tendon reflexes
- Personality change: Mild; 2 patients
- Disease progression
- Slow to Rapid
- Disease duration: Mean 9 years; Range 1 to 29 years
- ALS + FTD: 1 patient
- Laboratory
- EMG: Normal to Moderate denervation
- SSEP: Normal in 1 patient
- Muscle biopsy: Rare atrophic muscle fibers
- Autopsy: Lower motor neuron loss
- Animal model: 'pale tremor' (plt) mouse
● Profilin-1 (PFN1)
- Epidemiology
- < 30 patients
- 3% of FALS
- 0.2% of SALS
- Genetics
- Mutations
- Actin binding region
- A20T; C71G; ?T109M; M114T; M114V; E117G (also in controls); G118V; R136W; Q139L
- 2 singleton patients with no family history
- Mutations
- PFN1 protein
- Converts monomeric G-actin to filamentous F-actin
- Formin dependent
- Axonal integrity & Axon transport
- Nucleotide exchange factor
- Binds ATXN2 & VCP
- Mutant proteins
- May form ubiquitinated aggregates: Increased p62 & ubiquitin in neurons
- Alter growth cone morphology
- Altered actin polymerization into filaments
- Altered autophagy & mitochondrial distribution
- Converts monomeric G-actin to filamentous F-actin
- Clinical: Similar to SOD1
- Onset age: 4th to 7th decade; Mean > 50 years
- Onset region
- Limbs: All patients, Especially distal legs
- Bulbar: No patients
- Lower motor neuron: Most defecits
- Upper motor neuron: Few deficits; Tendon reflexes in arms may be brisk
- Respiratory: Late in disese course
- Independent walking: Often lost in 1st 2 years
- Cognitive: Normal
- Course: Progressive; Death < 3 years
- Brain pathology
- Motor neurons: Loss; TDP43 & p62 inclusions
- NO PFN1 inclusions
- Corticospinal tracts: Moderate myelinated axon loss
● V-erb-B2 Avian erythroblastic leukemia viral oncogene homolog 4 (ErbB4; HER4)
- Epidemiology: Japanese, Chinese (0.67% of sALS; 1.2% of fALS) & Canadians
- Genetics
- ErbB4 protein
- sALS: ERBB4 immunoreactivity reduced in spinal cord
- Clinical
- Onset
- Age: Adult; 36 to 70 years
- Arm involvement
- Bulbar: 2 patients
- Lower motor neuron
- Weakness: Limbs
- Respiratory insufficiency
- Upper motor neuron
- Cognitive: Normal or ALS + FTD in 1 patient
- Progression
- Slow, usually
- Death: After 1.5 to 20 years
- Onset
● D-amino acid oxidase (DAO)
- Epidemiology: 1 family
- Genetics
- Mutation: R199W
- May be schizophrenia
locus
- DAO protein
- Tissues: Liver, Kidney, Brain (Neurons & Astrocytes)
- Peroxisomes
- Homodimer
- Flavoenzyme
- Oxidizes: D-serine, D-alanine, D-proline
- Mutation effect: ? Dominant negative
- Clinical
- Onset: Hand weakness & wasting
- Upper motor neuron features
- Lower motor neuron involvement
- Bulbar signs
- Disease progression: Rapid; 1 to 2 years
- Age at death: Mean 44 years; Early 40's in 75%; Range 42 to 73 years
- Pathology in obligate carrier
- Motor neuron loss: Cervical & Lumbar spinal cord
- Lateral corticospinal tract pathology
- Frontal & temporal lobes: Preserved
- TDP-43 in motor neurons: Localized to nuclei; No inclusions
- DAO activity in spinal cord: Reduced
● TAF15 RNA Polymerase II, TATA Box-binding protein-associated factor, 68-kD (TAF15)
- Epidemiology: 6 sporadic patients; 0.5% of sporadic ALS
- Genetics
- Mutations
- Type: Missense;
- Location: Exons 13-16; M368T, G391E, R408C, G452E, G473E
- Allelic with: Chondrosarcoma, extraskeletal myxoid
- Mutations
- TAF15 protein
- Contains: Prion-like domain
- RNA recognition motif (RRM)-containing
- Location: Nucleus & Cytoplasm
- TET proteins (translated-in-liposarcoma, Ewing�s & TAF15)
- Mutant protein: Cytoplasmic foci
- Clinical: ALS, Adult onset
- Onset age: 5th to 7th decade
- Weakness: Distal & Proximal; Bulbar; Asymmetric
- Tendon reflexes: Brisk
- Sensation: Normal
- ALS + FTD: 1 patient
- EMG: Denervation
● Ewing sarcoma Breakpoint 1 (EWSR1)
- Epidemiology: 3 sporadic ALS patients
- EWSR1 protein
- Location: Nucleus; Cytoplasm, punctate or granular, in ALS neurons
- Contains: Prion-like domain
- Aggregation prone: Like FUS, TDP-43, TAF15
- Genetics
- Mutations: Missense; G511A, P552L
- Clinical: ALS
- Onset ages: 35 & 50 years
● Tubulin α-4A (TUBA4A)
- Epidemiology: 12 patients
- Genetics
- Mutations: Val7Ile; G34V; T145P; R215C; R320C; R320H; The349Ser; A383T; W407X; Asp438Asn; K430N
- TUBA4A protein
- Expression: Ubiquitous; High in brain
- Component of Microtubules
- See
- Mutations
- Destabilize microtubule network
- Reduced repolymerization capability
- Clinical: ALS
- Onset ages: 48 to 71 years
- Spinal-onset
- Upper & Lower motor neuron signs
- Frontal dementia: 3 patients & 1 1st degree relative
- Disease duration: 1 to 7 years
● TANK-Binding Kinase 1 (TBK1)
- Epidemiology
- Frequency: 1% to 3% of ALS & ALS/FTD; > 40 families
- Common in Belgium
- Genetics
- Mutations
- Types
- Loss-of-function
- Missense: E696K
- Effects: Loss of function; ? Dominant gain of function
- Heterozygous
- Types
- Penetrance
- Incomplete
- Family history: Often negative
- Allelic disorders: Different syndromes in same family
- Increased TBK1 copy number: Glaucoma (Primary open angle; Normal tension)
- PLS/Dementia
- Primary progressive aphasia
- Parkinsonism, atypical: Cortico-basal syndrome
- Encephalopathy, acute, infection-induced (herpes-specific), susceptibility to, 8 (IIAE8)
- Mutations
- TBK1 protein
- Kinase family
- Serine/Threonine
- IKK (IκB)
- Autophagy
- Efficient cargo recruitment in autophagy
- Degradation of protein aggregates
- Regulates selective autophagy
- Co-localizes with & phosphorylates OPTN & SQSTM1 in autophagosomes
- NF-κB pathway
- Related to: Innate immunity, type I IFN response
- Inhibits RIPK1
- Kinase family
- Clinical
- ALS syndrome
- Onset
- Age: Mean 60 years; Oldest 80 years; 9 years later than c9orf72
- Common: Legs or Arms
- Bulbar in 15% to 40%
- Bulbar involvement: 87%; Dysarthria; dysphagia
- Upper motor neuron
- Tendon reflexes: Brisk
- Spasticity: May be asymmetric
- Lower motor neuron
- Fasciculations
- Muscle atrophy
- Onset
- Fronto-Temporal dementia
- Cognitive impairment in 50%
- Language impairment (Aphasia)
- Presenting feature in 60%
- Extrapyramidal features: Hypokinesia; Tremor; Rigidity
- Course
- Survival: Mean 42 months; Range 1 to 9 years
- Age at death: Mean 65 years; Range 50 to 77 years
- ALS syndrome
- Laboratory
- EMG
- Denervation, Active & Chronic
- May be normal
- Brain MRI: Atrophy, often temporal & asymmetric
- FDG-PET: Asymmetric hypoperfusion or glucose hypometabolism
- Pathology
- Brain atrophy: Frontal & Temporal
- TDP-43 & p62 inclusions: Neurons & Glia
- Motor neuron loss
- EMG
- TBK1 variant: PLS/Dementia
140
- Epidemiology: 1 Spanish family, 6 patients
- Genetics
- Inheritance: Dominant
- Mutation: Arg573Gly
- Clinical
- Onset age: 60 to 66 years
- Dementia: 67%
- Behavioral changes
- Aphasia, Non-fluent
- Memory deficits
- PLS syndrome: 33%
- Distribution: Bulbar ± Spinal
- Course: Slow progression
- May occur without dementia
- EMG: No denervation
● Cyclin F (CCNF)
- Epidemiology: 72 patients; Diverse geography; 0.9% of ALS
- Genetics
- Mutations: 43 diffferent; Missense, mostly; S3G, K97R, T181I, S195R, R392T, S509P, T543I, S621G (Segregates), E624K, I772T
- Similar locus to: ALS-FTD 16p12
- CCNF protein
- F-box
- Component of E3 ubiquitin�protein ligase complex (SCFCyclin F)
- Genome stability
- Regulates: Deoxyribonucleotide triphosphate levels, Centrosome duplication, Spindle formation
- Targets & regulates levels of SCFcyclin F substrates
- Effects VCP activity in cytoplasm
- Clinical
- Onset age: Mean 53 years; Range 42 to 66 years
- Syndromes
- ALS alone
- Most common presentation
- Limb or Bulbar (10% to 27%) onset
- UMN + LMN signs
- Asymmetric
- PLS syndrome: 1 patient
- FTD-ALS syndrome: 1 patient
- Disease duration: 3 to 4 years (43 months)
- ALS alone
- Laboratory
- TDP 43 brain pathology: 1 patient
● CYLD Lysine-63 Deubiquitinase (CYLD)
- Epidemiology: European Australian family,
- Genetics
- Mutation: Missense; c.2155A>G, p.M719V
- Allelic disorders
- Brooke-Spiegler syndrome
- Cylindromatosis, familial
- Trichoepithelioma, multiple familial, 1
- Brooke-Spiegler syndrome
- CYLD protein
- Clinical
- Onset age: 30 to 62 years
- ALS
- Weakness: Flaccid paralysis
- Bulbar
- Dysarthria
- Dysphagia
- Jaw jerk: Present
- Tongue: Wasting; Fasciculations
- Sensory: Normal
- Dementia: Memory loss; Behavior problems
- Course: Death 5 to 12 years after onset
- Other occasional: Parkinson disease; Paget
- Pathology
- Fronto-Temporal atrophy
- Tauopathy: Cytoplasmic neuronal reactivity
- phospho-TDP-43 Cytoplasmic neuronal inclusions
- Hippocampal neurofibrillary tangles
- Glial CYLD-immunoreactivity in white matter
|
● Huntingtin (HTT)
|
HTT (PolyQ) aggregates Not in nuclei; Motor cortex 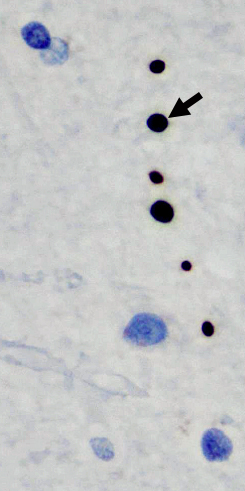 From: R Bucelli; R Hickman (Columbia, NY)
|
● SS18-like gene 1 (SS18L1; CREST)
- Epidemiology: 2 patients
- Genetics
- Possible Mutations: Q388X; I123M; c.660_668del, p.Gln222_Ser224del; c.790G>A, p.Ala264Thr
- SS18L1 protein
- Location: Nucleus
- Calcium regulated transcriptional activator
- Subunit of neuron-specific chromatin remodeling complex (nBAF)
- Neural outgrowth
- Interacts with: FUS
- Clinical
- Onset age: Adult to 8th decade
- ALS syndrome
● Annexin A11 (ANXA11)
- Epidemiology: 20 patients; 1% of FALS, 1.7% of SALS
- Genetics
- Mutations: Missense; P36R, G38R, D40G (Common European founder), D40Y, G175R, G189E, R235Q,R346C, G491R
- Penetrance: Incomplete
- Mutations also found in sporadic ALS patients
- Allelic disorders
- ALS23
- Multisystem proteinopathy 6 (MSP6; IBMWMA)
- OPMD syndrome, child onset
- Disease susceptibility
- R230C polymorphism associates with Autoimmune disorders & Sarcoidosis
- ? Sarcoidosis susceptibility (SS3)
- ANXA11 protein
- Calcium binding
- Subcellular location: Nucleus & cytoplasm (vesicle-like structures)
- Binds to: Calcyclin; Phospholipids, RNA granules, Lysosomes
- Roles
- Intracellular trafficking
- Vesicle transport
- RNA: Granules & Transport, Long distance with interaction with lysosomes
- Breast cancer & other malignancies: Increased expression
- 56-kD antigen recognized by sera from patients with autoimmune diseases
- Clinical: ALS
- Onset
- Age: Late; Mean 67 years; RAnge 37 to 71 years
- Bulbar (80%)
- Weakness
- Bulbar: Dysarthria; Dysphagia
- Limbs
- Age at death (D40G mutation): 71 to 85 years
- Cognitive impairment (FTD): Some patients
- Onset
- Laboratory
- Brain
- Neurons: Inclusions
- Cytoplasmic ANXA11, Phospho-TDP-43 & p62
- Morphology: Skein-like, Tubular-shaped, Filamentous & Complex basket-like
- Brain locations: Motor cortex; Occipital lobe
- Astrocytes: Increased calcyclin
- Neurons: Inclusions
- EMG: Denervation
- Brain
- ANXA11 variant syndrome: Multisystem proteinopathy 6
174
- Nosology: Inclusion body myopathy + Brain white matter abnormalities (IBMWMA)
- Epidemiology: Brazilian & Greek families, 18 patients
- Genetics
- Inheritance: Dominant
- Mutation: D40Y (Also found in ALS patient)
- Clinical
- Onset age: Usually 5th or 6th decade
- Weakness
- Asymmetric
- Legs > Arms
- Proximal
- Axial: Head ptosis; Abdominal; Scapular winging
- Gastrocnemius: Medial
- Face: Ptosis, mild
- Gait disorder
- Course: Slow progression
- ALS: 2 patients
- Spinal onset
- Course: Rapid progression
- Fronto-Temporal Dementia (20%)
- No Paget disease of bone
- Laboratory
- Muscle pathology
- Rimmed vacuoles
- Aggregates: Annexin A11, TDP-43, p62, Myotilin
- No inflammation
- Ultrastructure: Myofibrillar Δ Autophagic material
- EMG
- Myopathic ± Chronic denervation: May be presymptomatic
- Fasciculations: ALS syndrome patients
- Muscle MRI
- Arms: Anterior (Biceps) involvement
- Thigh: Adductors; Posterior (Semitendinosus, Semimembranosus)
- Leg: Gastrocnemius, medial
- Paraspinous
- Brain imaging
- White matter damage: Corticospinal tracts; Frontal subcortical
- Muscle pathology
● Glycosyltransferase 8 domain-containing protein 1 (GLT8D1)
- Epidemiology: 14 patients
- Genetics
- Mutations: Exon 4; R92C (Common; Short survival), I70T (Long survival), G78W (Long survival), A82E, I87N
- Additional ARPP21
mutation (P529L): Shorter survival
- GLT8D1 protein
- Glycosyltransferase
- Widely expressed: Neurons; Golgi localization signal
- Mutated proteins: Reduced glycosyltransferase activity
- Clinical
- Onset ages: 33 to 66 years; Average 49 years
- Weakness: Limb or Bulbar
- Disease duration: 6 to 101 months
- Heterogeneous category of: Non-SOD linked; Adult onset; Hereditary ALS syndromes
- Comprises 30% to 50% of dominant ALS syndromes
ALS SYNDROMES: RECESSIVE
- ALS 2: Childhood-onset ALS: Long survival; Usually Recessive
● Alsin (ALS2); Chromosome 2q33.1; Recessive
- Genetics
16
- Mutations
- Disease related: Carboxyl-terminal
- ALS2: Deletions in exon 3 or 4
- Disrupt reading frame
- Allelic with
- Juvenile PLS: Caused by different mutation in same gene
- SPG, Infantile onset
- Juvenile ALS + Dystonia
- Mutations
- Protein
40
- Contains 3 guanine-nucleotide exchange factor (GEF) domains for GTPases
- Localization
- Cytoplasmic face of endosomal membranes
- Requires amino-terminal "RCC1 (regulator of chromatin condensation)-like"" GEF domain
- Possible functions
- ? Involved in membrane-proximity activities of small GTPases
- ? Related to vesicle transport & intracellular trafficking
- Tissue Expression
- Neurons in brain & spinal cord
- Low abundance in all tissues
- Mutants
- All have short half-life
- Rapidly degraded by proteasomes
- Clinical features (Type 3)
- Onset age: Mean 6.5 years; Most often < 10 years; Occasional 3rd decade
- Spasticity: Face; Limbs (Paraplegia)
- Pseudobulbar affect
- Amyotrophy: Mild; Distal; Legs > Arms; Increases with age
- Sensory & Bladder: Normal
- Progression: Very slow; Reduced Walking after age 40
- Laboratory
- CSF protein: Normal
- Nerve conduction velocities: Normal
- Nerve biopsy: Mild Reduction in number of large & small axons
- ALS2 variant syndrome: Juvenile ALS + Dystonia
110
- Epidemiology: 8 patients, 3 families
- Genetics
- Inheritance: Recessive
- Mutations: Nonsense; Fameshift; Splice
- Clinical
- Onset age: 1 to 3 years
- UMN
- Spastic diplegia
- Dysarthria
- Gait disorder
- Tendon reflexes: Brisk; ankle clonus
- Cortical
- Developmental delay
- Emotional lability
- Weakness: Generalized
- Muscle wasting: Legs > Arms; Distal > Proximal
- Dystonia: Generalized
- Skeletal: Microcephaly; Scoliosis; Limb contractures
- Other
- Ataxia
- Anarthria
- Course: Bedbound at 5 to 19 years
- Normal: Hearing; Eye movements
- Laboratory
- Muscle pathology
- Small angular muscle fibers
- Fiber type grouping
- NCV: Normal
- Muscle pathology
- Genetics
16
- ALS 5: Childhood-onset
● Spatacsin;
Chromosome 15q21.1; Recessive
- Epidemiology
- 10 families
- ? Most common form of recessive ALS
- Genetics
70
- Spatacsin mutations
- Multiple
- Missense common; Some frameshift
- Many other families linked to similar locus
- Consanguinous families
- Allelic disorders
- Spatacsin mutations
- Spatacsin protein
- Clinical: Weakness, Arm atrophy & Spasticity in all limbs (Type 1)
- Onset age: Range 7 to 23 years; Mean 16
- Spasticity
- Limbs
- Face & Jaw
- Bulbar
- Hyperreflexia
- Plantar responses: Extensor
- Gait
- Amyotrophy & Weakness: Occasionally predominant
- Distal
- Hands & Feet
- Tongue
- Fasciculations
- Course
- Slowly progressive
- Survival: Duration 27 to 40 years
- Cognition: Normal
- Bladder: Normal
- Laboratory
- EMG: Denervation
- Positive sharp waves
- Fibrillations
- Fasciculations
- Nerve conduction
- CMAP amplitudes: Small
- SNAP amplitudes: Normal
- NCV: Normal
- Pathology
- Pyramidal tracts: Small
- Anterior horn cell loss
- Sural nerve: Reduced number of Myelinated axons
- Corpus callosum: Normal
- CSF protein: Normal
- EMG: Denervation
- Epidemiology
- ALS 12: Adult-onset
74
● Optineurin (OPTN);
Chromosome 10p13; Dominant or Recessive
- Epidemiology
- Japanese consanguinous, German, Italian & Dutch families
- 38 patients described
- 1% to 4% of FALS
- SALS: < 1%
- Genetics
- Mutations
- Japanese: Recessive or Dominant; Exon 5 homozygous deletion, Q398X homozygous, E478G heterozygous
- Other: All Dominant; Misense, Nonsense, Intronic
- Common location: C-terminal Ubiquitin-binding domain
- Possible mutation mechanism: Haploinsufficiency
- Allelic with: Primary open-angle glaucoma
- Most mutations: Single copy missense
- Possible mutation mechanism
- Overactivate OPTN-TBK1 pathway
- Cause increased OPTN & TBK1 phosphorylation & activity
- Mutation with either ALS or glaucoma
- 691_692insAG: Dominant or Recessive ALS
- Mutations
- OPTN protein
- Interacts with: Huntingtin
;
Transcription factor IIIA;
RAB8
- Colocalises with SOD1- and TDP-43-positive inclusions in ALS
- Binds to ubiquitinated receptor-interacting protein
- Negatively regulates TNF-α induced activation of NF-κB
- Autophagy-related
- Interacts with: Huntingtin
- Clinical
- Onset
- Age: Range = 24 to 83 years; Mean = 62 years
- Weakness: Legs > Bulbar, Arms
- Weakness
- Arms
- Legs
- Asymmetric
- Respiratory: Late in course
- Bulbar
- Some patients
- Dysphagia; Dysarthria; Tongue fasciculations
- Tendon reflexes: Normal or Increased
- Spasticity: Prominent in 70%
- Progression: Variable
- Range: 0.75 to 25 years
- Mean: 5 years
- Rapid progression: Q165X; Q454E; K557T; p.Lys440Asnfs*8
- Fronto-Temporal Dementia: Few patients
- No glaucoma
- Onset
- Laboratory
- EMG: Diffuse denervation
- Pathology
- Spinal cord: Loss of myelin & Axons
- OPTN cytoplasmic inclusions
- Epidemiology
- ALS 16: Childhood-onset
81
● Sigma-1 Receptor (SIGMAR1; σ1R);
Chromosome 9p13.3; Recessive
- Epidemiology: Eastern Saudi Arabian family
- Genetics
- Mutation: Homozygous; Missense; E102Q
- Allelic disorders
198
- ALS, Adult onset, Sporadic
- Parkinsonism-ALS, Adult onset,
- Distal HMN (dHMN)
- ? FTD-MND
- Similar locus to: Distal SMA2 (Jerash type, DSMA 2; HMNJ)
- SIGMAR-1 protein
- Type II membrane protein: Short cytosolic N-terminal tail 180
- Cell location: Endoplasmic reticulum (ER)
- Expression areas: Ubiquitous, High in Motor neurons
- Sub-cellular: Post-synaptic cholinergic densities (C-terminals)
- Chaperone
- Ligand: Neurosteroids, Psychostimulants, Dextrobenzomorphans, Dimethyltryptamine
- Functions
- Ion channel modulation: Through interaction with K+ channels & Inositol 1,3,5-triphosphate receptors (IP3Rs)
- Lipid transport
- Neuronal cell differentiation
- ER stress response
- Regulates: Axon elongation; Tau phosphorylation
- Mutation effect: Abnormal subcellular distribution of SIGMAR-1
- Clinical
- Onset
- Age: 1 to 30 years
- Spasticity: Legs
- Spasticity
- Legs > Arms
- Tendon reflexes: Brisk
- Weakness
- Hand & Forearm muscles: Especially extensors & triceps
- Normal: Sensation; Cognition; Bladder
- Progression: Often to wheelchair by 3rd decade
- Onset
- Laboratory
- EMG: Denervation in limb muscles; Motor units large
- NCV: Normal velocities
- Brain MRI: Normal
- SIGMAR1 Variant syndrome, Possible: Frontotemporal lobar degeneration-Motor neuron disease
- Inheritance: Dominant
- Epidemiology: Australian families
- Genetics
- 3' UTR point mutations: Some may be polymorphisms, also occur in normals
- Some patients & familes: Pathogenic mutation may be another gene, c9orf72 expansion
- SIGMAR1 Variant syndrome: Distal hereditary motor neuropathy (HMNR2; dHMN; DSMA2)
122
- Epidemiology: Chinese (3 patients), Portugal & Afghan families
- Genetics
- Inheritance: Recessive
- Mutations: Deletion, or Splice
- c.151+1G>T: Produces shortened protein (σ1R31_51del); Homozygous
- c.561_576del (Exon 4; Stop)
- Exon 4 deletion
- Similar locus to: Distal SMA2 (Jerash type, DSMA 2; HMNJ)
- Clinical
- Onset age: Range 5 to 12 years
- Weakness
- Distal
- Legs > Arms: Foot drop
- Symmetric
- Wasting: Distal; Legs > Arms
- Tendon reflexes
- Ankles: Absent
- Knees: Increased or Reduced
- Plantar reflex: Extensor
- Feet: Pes equinovarus; Hammer toes
- Sensory: Normal
- Course: Slow progression, more rapid during adolescence
- Laboratory
- NCV
- Velocities: Borderline (32 to 52 M/s); May vary among nerves
- CMAPs: Amplitudes reduced; Temporal dispersion in some
- SNAPs: Normal
- EMG: Distal denervation; Large motor units
- Sural nerve: Normal
- Brain MRI: Normal
- NCV
- ALS
188
● Ring finger Protein 13 (RNF13);
Chromosome 3q25.1; Recessive
- Epidemiology: 1 family; 3 patients
- Genetics
- Mutation: Homozygous; Intron; c.195+1G>A
- Allelic disorder: Developmental and epileptic encephalopathy 73 (DEE73)
- RNF13 protein
- Mediator of endoplasmic reticulum stress response & apoptosis
- Interactions: ERN1
; JNK
- Neurite outgrowth: Stimulation
- Mutation effect: Abnormal RNF13 splicing
- Clinical
- Onset ages: 4th & 5th decade
- Weakness: Arms, then legs
- Upper motor neuron: DTRs increased; Leg spasticity in 2 patients
- Extrapyramidal: Bradykinesia; Tremor
- Sensation: Normal
- Course: Slow progression
- Laboratory
- EMG: Denervation, diffuse
- Brain MRI: White matter - Periventricular hyperintensities, Corpus callosum atrophy
- ALS: Childhood-onset
64
● Chromosome 6p25, 21q22; Recessive- Epidemiology: 1 Utah family, 4 patients
- Clinical
- Onset age: 4 to 10 years
- Ptosis: Mild
- Weakness & Atrophy
- Distal
- Arms & Legs
- Symmetric
- Respiratory failure: Late in course, 1 patient
- Bulbar & Cranial
- Speech: Dysarthric; Spastic
- Dysphagia
- Face weakness, mild
- Upper motor neuron
- Spasticity: Legs > Arms
- Tendon reflexes: Brisk in legs ± arms
- Plantar response: Upgoing or absent
- Sensory loss: Mild; Distal; Vibration
- Gynecomastia: 1 patient
- Progression: Slow; Loss of ambulation after decade
- Laboratory
- NCV
- Normal velocities
- CMAPs: Reduced amplitude in legs
- SNAPS: Reduced amplitude
- EMG: Denervation, Distal
- Nerve biopsy: Minimal loss of sensory axons
- Muscle biopsy: Grouped atrophy; Fiber type grouping
- NCV
- Recessive ALS: Other types
- Clinical syndrome: Weakness & severe spasticity, esp. in legs (Type 2)
- Symptoms in lower limbs
- ? form of familial spastic paraplegia
- Clinical syndrome: Weakness & severe spasticity, esp. in legs (Type 2)
BULBAR MOTOR NEURON SYNDROMES: Hereditary & Other
|
Brown-Vialetto-van Laere 1: SLC52A3; 20p13; Recessive 2: SLC52A2; 8q24; Recessive 3: UBQLN1; 9q21; Dominant Bulbar ALS Fazio-Londe: SLC52A3; 20p13; Recessive Kennedy's Syndrome: Androgen Receptor ; Xq12; Recessive Madras motor neuron disease: Sporadic Spino-bulbar muscular atrophy: Dominant Worster-Drought |
Fazio-Londe
● SLC52A3 (RFT2; c20orf54)
- Definition
- Progressive bulbar palsy of childhood
- Disorder of bulbar motor neurons
- Genetics
- Allelic with: Brown-Vialetto-van Laere 1 (BVVLS1)
- Clinical features
- Onset: 1 to 12 years
- Cranial nerves
- Ptosis
- Facial weakness
- Dysphagia
- Normal hearing
- Respiratory stridor
- Hyperreflexia
- Subtypes
- Dominant: Rare
- Recessive: Early onset
- Respiratory failure
- Rapid progression: Death < 2 years from onset
- Recessive: Later onset
- Less respiratory involvement
- Bulbar: Dysarthria; Dysphagia
- Facial weakness
- Slow progression
- Laboratory
- CSF: Normal
- Serum CK: Normal
- EMG: Denervation; Large amplitude Motor unit potentials
- Neuropathology
- Loss of neurons from motor cranial nerve nuclei
- Occasional: Anterior horn cell loss; Cerebelar involvement
- Similar to: Brown-Vialetto-van Laere
- But no hearing loss
Brown-Vialetto-van Laere 1 (BVVLS1)
● SLC52A3 (RFT2; c20orf54)
- Nosology: Progressive Bulbar Palsy with Sensorineural Deafness
- Epidemiology
- 50% of patients: Sporadic
- Sex associations
- Female predominant 60% to 75%
- Familial cases: Many female
- Males: Often have more severe course
- Family origin: Arabic; European; Pakistani
- Genetics
- Mutation types: Deletion; Missense; Stop
- c.1325_1326delTC; P28T; E36K; G54R; E71X; E71K;
I75T;R132W; Y213X; F224L; V413A; L442RfsX35; F457L - Allelic with: Fazio-Londe
- SLC52A3 (c20orf54) protein
- Transmembrane
- Riboflavin transporter
- Clinical features
- Onset
- Age
- Common: 1st or 2nd decade; Often < 8 years
- Range: Infancy to early 3rd decade
- May be variable within families
- Early development: Normal
- Hearing loss: Often precedes motor syndrome
- Gait disorder
- May follow episode of viral infection
- Age
- Cranial nerves
- Deafness (100%)
- Bilateral
- Sensory-Neural
- Weakness: Especially 7, 9,and 12
- Facial: Bilateral
- Bulbar
- Tongue: Wasting & Fasciculations
- Dysphagia
- Eye
- Optic atrophy (90%)
- Most: Normal eye movements
- External ophthalmoplegia: 1 patient
- Deafness (100%)
- Motor
- Weakness
- Especially: Arms & Hands, Neck (Extension) & Trunk
- Respiratory failure: More severe cases
- Gait disorder
- Dysphagia: Some patients
- Tone: Reduced
- Muscle wasting
- Weakness
- Tendon reflexes: Normal or Reduced
- Sensory examination: ?
- CNS
- Truncal ataxia: Some patients
- Normal intelligence
- Skeletal: Contractures, distal; Scoliosis
- Other: Occasional features
- Congenital heart disease
- Disease course
- Irregularly progressive
- Early onset: More rapid course (6 to 18 mo)
- Death
- Death in more severe patients: Respiratory failure; 1 to 2 years
- Some patients live through 6th decade
- Treatment: Riboflavin replacement, High dose (100mg to 500 mg per day)
- Onset
- Laboratory
- Brainstem auditory responses: Absent
- EMG: Denervation
- NCV: Motor velocities: Normal SNAP amplitudes: Reduced or absent
- Acyl carnitine profiles: Abnormal; Similar to MADD deficiency
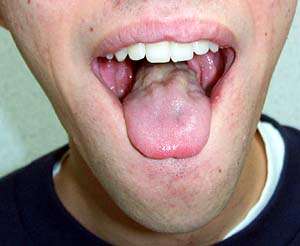
 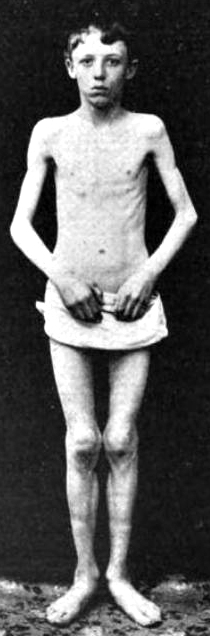 |
Brown-Vialetto-van Laere 2 (BVVLS2)
● SLC52A2 (RFT3; GPR172A; RFVT2)
- Nosology
- Progressive Bulbar Palsy with Sensorineural Deafness
- Childhood Neuronopathy
- Epidemiology: 3 families, Scottish, Italian & Lebanese
- SLC52A2 genetics
- Mutations: Gly306Arg, Homozygous; Ser52Phe, Gly419Ser; Leu123Pro, Leu339Pro
- Allelic disorder: SCAR3 (SCABD2)
- SLC52A2 protein
- Riboflavin transporter
- Brain: High expression
- Clinical
- Onset
- Age: 2 years
- Motor: Gait disorder; Exercise intolerance; Dysphonia
- Hearing loss: 2 to 6 years
- Hearing loss: Sensorineural
- Weakness
- Bulbar
- Arms & Hands > Legs
- Axial: Neck weakness; Hypotonia
- Tongue fasciculations
- Respiratory failure: 2nd decade
- Tendon reflexes: Absent
- Sleep: Hypoventilation
- Optic atrophy: Reduced visual acuity
- Scoliosis
- Hypermetabolic syndrome
- Course: Progressive; Early death in some patients
- Treatment: Riboflavin replacement, High dose (100mg to 500 mg per day)
- Onset
- Laboratory
- Polyneuropathy: Axonal; Sensory & Motor
- BAER & VER: Abnormal
- Acyl carnitine profiles: Abnormal; Similar to MADD deficiency
- Carnitine deficiency
Brown-Vialetto-van Laere 3 (BVVLS3) 94
● Ubiquilin 1 (UBQLN1)
- Epidemiology: 1 Italian female singleton patient
- Mutated UBQLN1 protein
- Induces cytosolic mislocalization of TDP-43
- In aggregate forms that co-localize with the UBQLN1
- Genetics
- Mutation: Missense, E54D; Heterozygous
- Other disease association: Intronic variant (UBQ-8i) related to Alzheimer disease
- Clinical
- Onset: Juvenile
- Motor neuron disease: Bulbar palsy
- Hearing loss: Sensorineural
Madras Motor Neuron Disease 61
● Sporadic; Occasional recessive inheritance
- Epidemiology
- Common in South India
- Family history of similar disorder: 2%
- Male = Female
- Genetics
- A8302G mutation in mt-tRNALys
100
- Homoplasmic in 7% of Madras patients
- High levels (82%) also in some unaffected family members
- No mutations in: SLC52A1, SLC52A2, SLC52A3, c9orf72
- A8302G mutation in mt-tRNALys
100
- Clinical
- Onset
- Age: Juvenile; Mean 16 years, Range 1st to 5th decade
- Hearing loss: 53%
- Arm weakness: 38%
- Cranial nerve
- VII: Facial diplegia, Lower > Upper
- VIII: Deafness, Sensorineural (30% to 80%)
- IX to XII: Bulbar involvement (40%)
- Dysphagia
- Dysarthria (Hoarseness)
- Tongue: Weakness; Fasciculations
- Lower motor neuron signs (85%)
- Weakness
- Limbs: Arms > Legs
- Asymmetric
- Diffuse: Distal > Proximal
- Wasting
- Fasciculations: Limbs & Face
- Weakness
- Pyramidal signs (80%)
- Reflexes
- Tendon reflexes: May be brisk in legs
- Plantar: May be extensor (50%)
- Sensory: Normal
- Other: ? Minipolymyoclonus related to fasciculations
- Course
- Slowly progressive
- Most remain ambulatory
- Onset
- Laboratory
- Electrodiagnostic
- EMG: Active & Chronic denervation in cranial, proximal & distal muscles
- CMAPs: Small amplitude
- SNAPs: Normal
- Pathology: 1 patient
- Anterior horn cells: Reduced
- Ventrolateral column: Axonal loss
- Other cranial nerve nuclei involved: 3, 5, 6, 7, 10
- Cochlear nuclei: Neuronal loss
- Other tracts involved: Spinocerebellar & Spinothalamic
- Other regions of CNS also involved
- Cellular: Microglial reaction; No cytoskeletal pathology
- Electrodiagnostic
- Differential diagnosis: Similar to
- Variant: Madras MND + CNS features (13%)
33
- Epidemiology: India
- Family history
- Positive: 29%, More frequent than typical MMND
- Clinical
- Onset age: Younger than typical Madras MND
- Bulbar dysfunction (100%): More common than typical Madras MND
- Optic atrophy (100%)
- Cerebellar disorder: Ataxic gait
- Pyramidal signs
- Minipolymyoclonus
Worster-Drought syndrome: Congenital suprabulbar paralysis
- Inheritance patterns
- X-linked
- Autosomal dominant with reduced penetrance
- Clinical
- Course
- Onset: Congenital
- Age at diagnosis: 6 years
- Non-progressive
- Male > Female: 3:1
- Familial cses: 6% to 21%
- Bulbar paralysis
- Abnormal tongue & lip movement (98%)
- Brisk jaw jerk (90%)
- Airway obstruction: Especially neonatal presentation
- GI: Dribbling; Poor nutrition; Aspiration; G-E reflux
- Speech: Dysarthric
- Spastic tetraparesis (91%): Mild
- Cortical
- Language disorder: Expressive > Receptive
- Learning difficulties (81%)
- Neuropsychiatric disorders (41%)
- Epilepsy (28%): Neonatal; Febrile; epilepsy
- Congenital defects (61%)
- Contractures: Jaw & Other
- Palatal
- Micrognathia
- Fused teeth
- Eye movements: Abnormal (13%)
- Course
- Laboratory
- CNS Imaging
- Abnormal in 32%
- Perisylvian polymicrogyria: Unilateral or Bilateral
- Swallowing studies: Abnormal, especially in oral & pharyngeal phases
- CNS Imaging
Cortical Suprabulbar palsy
- Foix-Chavany-Marie syndrome: Faciopharyngoglossomasticatory diplegia
- Loss of voluntary control: Preserved automatic function
- Anatomy: Bilateral persylvian lesions
Multisystem disorders
- Hexosaminidase A: GM2 Gangliosidosis, Late Onset
● β-Hexosaminidase A (HEXA)
;
Chromosome 15q23; Recessive
- Nosology: Tay-Sachs, Late onset (LOTS)
- Genetic features
- Gly269Ser substitution in α chain
- Most common mutation in adult form
- Usually in compound heterozygosity with null mutation
- Most common mutation in adult form
- Other mutations with milder disease
- Missense: Arg499His; Arg499Cys; Arg504His; Arg504Cys
- Splice site: IVS7-7G
- General: Chilhood + Adult mutations
- Other mutations (null): More severe disease
- Exon 11: 4-bp insertion
- Exon 12: G to C mutation in splice junction
- Exon 1: A to T in initiation codon
- Exon 11: 4-bp insertion
- Misdiagnosis
- Pseudo-deficiency alleles: Arg247Trp; Arg249Trp
- False negative: B1 variant; Abnormal reactivity to GM2 but not artificial substrate
- Gly269Ser substitution in α chain
- HEXA protein
- Location: Lysosome enzyme
- Function
- Breakdown of gangliosides
- Hydrolyzes GM2 ganglioside to GM3
- Disease syndromes: < 10% of normal Hex A activity
- Later-onset syndromes: Higher Hex A activity
- Clinical features
- Onset: Cramping in legs
- Weakness
- Proximal > Distal
- Selective: Triceps; Quadriceps
- Symmetric
- May be only sign, or present with CNS features
- Spontaneous activity
- Cramps
- Fasciculations
- Cerebellar
- Gait Ataxia
- Dysarthria
- Nystagmus
- Tremor
- Cortical function
- Dementia
- Manic-Depressive-like disorder
- Easy startle
- More common with neuropsychometric testing: Subcortical
- Deficits: Attention, Processing speed, Executive function, Memory
- May be normal
- Sensory: Unusual
14
- Paresthesias: Distal; 2nd decade onset
- Loss: Mild; Pansensory; Distal legs
- Tendon reflexes: Normal, Reduced in legs, or Brisk
- Corticospinal: Some patients
- Spasticity
- Bladder dysfunction
- Movement disorder: Dystonia & Choreoathetosis
- Cranial nerves: Spared
- Exacerbating drugs: Psychotropic medications (Haloperidol, Chlorpromazine, Risperidone)
- Lab
- MRI: Cerebellar atrophy common
- Nerve biopsy: Axon loss, Sensory & Motor
- Muscle biopsy: Denervation, Grouped atrophy, Pyknotic nuclear clumps
- NCV: Small amplitude CMAPs & SNAPs
- EMG: Large motor units; Fibrillations; CRDs
- Autonomic pathology: Membranous cytoplasmic bodies (MCBs) in neurons
- Experimental treatments
- Lower motor neuron phenotype also with Sandhoff disease
- Machado-Joseph
(esp Type I)
● CAG triplet repeats; Chromosome 14q32.12; Dominant- Clinical features
- Type I = Earlier onset (20 to 30 yrs) with longer CAG repeats
- Dystonia; Facial & Lingual Fasciculations; Bulging Eyes
- Cerebellar signs with distal amyotrophy in later onset (Types II & III)
- Allelic with Spino-Cerebellar Ataxia (SCA) III
- Type I = Earlier onset (20 to 30 yrs) with longer CAG repeats
- Clinical features
- Polyglucosan body disease
: Adult-form
● Glycogen branching enzyme (GBE1);
Chromosome 3p12.2; Recessive
- Epidemiology
- Frequency: 1 in 600,000 to 800,000
- Ashkenazi Jewish patients: Increased fequency; Carrier 1 in 48
- Non Ashkenazi Jewish: German, Italian, Latin American, Pacific Islander, Caucasian & Cambodian
- Genetics
- Mutations: Missense
- Ashkenazi Jewish patients: Tyr329Ser
- German & Italian: Arg515His & Arg524Gln
- Allelic disorders: GSD 4
- Mutations: Missense
- GBE1 protein
- Clinical features: CNS + Polyneuropathy
39
- Onset
- Age
- Adult: 20 to 71 years; Mean 51 years
- Other forms in childhood: Myopathy & Hepatic disease
- Neurogenic bladder
- Age
- Polyneuropathy
- Legs > Arms
- Motor: Weakness, Distal
- Sensory loss: Large fiber modalities (Vibration)
- Autonomic: Impotence; Orthostatic hypotension; Fecal incontinence
- Muscle
- Weakness (50%): Proximal
- Muscle MRI: Glutei; Semimembranosus; Biceps femoris; Gastrocnemius
- Myelopathy
- Spasticity
- Legs > Arms
- Gait disorder
- Tendon reflexes: Brisk in legs
- Plantar responses: Extensor
- Neurogenic bladder
- Spasticity
- Dementia & Cognitive (50% to 67%)
- Cortical & Subcortical
- Attention & Memory defecits
- Course
- Bladder Δ → Gait disorder → Cognitive decline
- Survival: Mildly reduced; Mean 70 years
- Atypical features in some patients
- Earlier onset
- Ataxia
- Parkinsonism
- Cardiomyopathy, dilated
- Course
- Progression: Slow
- Onset
- Laboratory
- MRI
- Cortical atrophy
- White-matter changes (Leukodystrophy): Periventricular; Subcortical
- Distribution: Bilateral; Symmetric; Occipital
- Cavitary changes: Internal capsule; Anterior perforated substance
- FLAIR: Hyperintense pial rim; T2 hyperintense mesencephalic �beak�
- Corpus callosum: Thin
- Diffuse atrophy: Brain & Spinal cord
- Nerve conduction studies: Neuropathy
- Axon loss
- Sensory & Motor involvement
- Serum CK: May be high
- Liver enzymes (AST & ALT): May be mildly high
- GBE activity: Reduced by 60% to 80%
- Muscle fiber pathology
- Basophilic aggregates
- PAS: Dense positive, α amylase resistant material
- MRI
- Nerve & CNS Pathology
- Dx by sural nerve or axillary skin biopsy
- Polyglucosan bodies
- Perivascular inflammation: Occasional
- Epidemiology
- Motor Neuronopathy with Cataracts & Skeletal abnormalities
● ALDH18A1 (P5CS); Chromosome 10q24.1; Dominant
- See SPG 9A (ALDH18A1)
- Clinical features
- Onset: 1 to 37 years
- Cataracts
- Weakness
- Distal arms & proximal legs
- Exacerbation during pregnancy; remission afterwards
- Distal arms & proximal legs
- Upper motor neuron signs
- Skeletal
- Short stature; Dysplastic skull base; Small carpal bones
- Disinhibition-Dementia-Parkinsonism-Amyotrophy complex (DDPAC; Fronto-Temporal dementia)
● Microtubule-associated protein tau (MAPT);
Chromosome 17q21.31; Dominant
- Allelic with
- Frontotemporal dementia with parkinsonism
- Frontotemporal dementia with motor neuron disease
- Progressive supranuclear palsy
- Tauopathy + Respiratory failure (Recessive)
- Pick disease
- Lower motor neuron disorder
- Parkinson-ALS
- Parkinson susceptibility
- Clinical
- Onset: Mean age 45 years; Alcoholism or Depression
- Dementia: Behavior & judgement > Language & praxis; Kluever-Bucy syndrome
- Disinhibition: Alcoholism; Hyperreligiosity; Excessive eating, sexual behavior
- Parkinsonism
- Amyotrophy
- Variability of symptoms among patients
- Pathology
- Superficial cortex: Spongiform changes
- Spinal cord: Motor neuron loss
- MAPT variant syndrome: Lower motor neuron syndrome
112
- Epidemiology: 1 family, 5 patients
- Genetics
- MAPT mutation: D348G
- Inheritance: Dominant
- Clinical
- Onset age: 6th & 7th decade
- Weakness
- Proximal: Arms
- Respiratory
- Tendon reflexes: Reduced
- Leg cramps
- Fatigue
- No: Frontotemporal lobar degeneration; Semantic dementia; UMN features
- Pathology
- α-motoneurons
- Loss
- Phosphorylated tau: Accumulation in surviving motor neurons
- Spinal anterior horns: Atrophy
- α-motoneurons
- MAPT variant syndrome: FTD + Motor neuron syndrome
- Genetics
- Mutation: L317M
- Inheritance: Dominant
- Clinical
- Dysarthria
- Tremor
- Amyotrophy
- Frontal signs
- Levodopa-resistant parkinsonism
- Supranuclear palsy
- Corticobasal degeneration
- Pathology
- Spinal motor neurons: Loss
- Tau inclusions
- Genetics
- MAPT variant syndrome: Parkinson-ALS
197
- Nosology: Brait�Fahn�Schwarz disease
- Epidemiology: 1 patient
- Genetics
- Mutation: Pro494Leu; Heterozygous
- Clinical
- Onset age: 6th decade
- Parkinsonism
- ALS syndrome: After Parkinsonism
- Weakness
- Fasciculations
- Upper motor neuron: Spasticity; Tendon reflexes brisk
- Speech: Dysarthric
- Course: Progressive over 2 years
- Laboratory
- NCV: CMAP amplitude, small
- EMG: Denervation, diffuse
- Allelic with
- MAPT variant syndrome: Tauopathy + Respiratory failure
- Epidemiology: 1 family, 2 patients
- Genetics
- Mutation: Homozygous; Ser352Leu
- Inheritance: Recessive
- Clinical
- Onset age: 3rd decade
- Respiratory failure, Acute
- Learning difficulty: Early onset; 1 patient
- Pathology
- Tau accumulation: In some neuron cell bodies
● Signal transducer and activator of transcription 5B (STAT5B)
- Epidemiology: 2 patients, 1 family
- Genetics
- Mutation: Glu315Ala
- STAT5B protein
- Signal transduction
- Transcription activator
- Clinical
- Growth retardation: Short stature
- Dysmorphic face: Ptosis
- Motor neuropathy
● Autosomal Recessive
- Epidemiology: 1 patient
- Clinical
- Weakness: Bulbar; Arms; Respiratory failure
- Ophthalmoplegia: Reduced gaze excursion, especially up; Supranuclear
- Progression: Over 1 year
- Laboratory
- EEG: Diffuse slowing
- CT: Atrophy of frontal lobe & midbrain
- Brain: Abnormal anterior horn & corticospinal tracts; Bunina bodies
● RBM28
- Epidemiology: Arab Moslem kindred
- Genetics
- Mutation: Missense; Leu351Pro
- RBM28 protein
- Component of snRNP complexes
- Ubiquitous expression
- Regulates ribosome biogenesis
- Mutant: Ribosome depletion; Structural abnormalities of rough endoplasmic reticulum
- Clinical
- Skin
- Hair loss
- Widely varying severity
- Skin biopsy: Absence of mature hair follicles; Reduced β-catenin
- Pigment
- Flexural reticulate hyperpigmentation
- Facial pigmented nevi: Multiple
- Subcutaneous fat: Loss
- Hair loss
- Motor
- Progressive decline
- Combined upper & lower motor dysfunction
- Onset: 2nd decade
- CNS
- Mental retardation: Moderate to severe
- Endocrine: Central hypogonadotropic hypogonadism
- Delayed or absent puberty
- Central adrenal insufficiency
- Gynecomastia
- Skeletal
- Short stature
- Microcephaly
- Hypodontia
- Kyphoscoliosis
- Ulnar deviation of hands
- Skin
- MRI
- Hypoplastic pituitary gland with preserved hypothalamus
- Normal basal ganglia & white-matter
● SCN1A
- Nosology: Epileptic encephalopathy, early infantile, 6 (EIEE6)
- Epidemiology: Rare epilepsy syndrome
- Genetics
- SCN1A protein
- Clinical
- Ataxia: Gait
- Seizures: Severe; Onset 2 to 6 months; Pharmacoresistant
- Psychomotor delay: Moderate to severe
- Orthopedic
- Misalignment
- Knee hyperextension
- Foot: Pes valgus
- Crouching
- Movement disorders
- Onset age: Adulthood
- Lingual dyskinesia
- Rigidity
- Dystonic gait
- Treatable with levodopa
- Tendon reflexes: Reduced (30%)
- Laboratory
- EEG: Normal then Generalized spike-wave activity
- NCV: Normal
- EMG: Motor neuronopathy
130
- Chronic denervation, especially > 4 years
- Motor units: Prolonged; High amplitude; Polyphasic
● von Willebrand Factor A Domain-containing Protein 1 (VWA1)
- Epidemiology: 34 patients, 23 families
- Genetics
- Mutations
- Types: Loss-of-function; Duplication & Deletion
- Common in Europeans: G25Rfs*74 10 bp repeat expansion (3 copies vs 2 in controls)
- Allelic disorder: Hemifacial microsmia, Dominant
, Missense mutation
171
- Mutations
- VWA1 protein
- Location: Basement membranes; Extracellular matrix
- Interactions: Collagen VI; Perlecan; FGF pathway
- Secreted
- Clinical
- Onset age: 1 to 54 years
- Weakness
- Legs: Anterior > Posterior; Foot drop
- Thigh: Anterior & Posterior
- Arms: Mild or None
- Proximal & Distal
- Myalgia (40%)
- Sensory: Normal
- Tendon reflexes: Reduced in 50%
- CNS: Normal
- Skeletal: Pes cavus (90%); Joint contractures (50%)
- Course: Slow progression
- Young onset: Upper motor neuron signs; Gait disorder
- Laboratory
- Muscle pathology
- Fiber type groups
- Fiber sizes: Varied
- Ultrastructure: Mitochondrial paracrystalline inclusions
- EMG: Fibrillations, distal; Myopathic, proximal
- NCV: Motor axon loss
- Muscle MRI
- Uniform pattern
- Thigh: Vasti
- Legs: Gastrocnemius (Medial > Lateral); Peroneal
- Serum CK: Normal to 1,600
- Muscle pathology
Motor Neuron Disease: Mouse models
- Wobbler
52
● Vacuolar protein sorting protein 54 (Vps54)
- Mutation: Missense L967Q
- Motor neuron disease
- Spermiogenesis defect
- Neuromuscular degeneration (nmd): SMARD1
- Muscle deficient: Mouse chromosome19
- Onset: Hindlimb weakness
- Peripheral nerve: Axonal degeneration
- Neurons: Ventral horn & Brainstem motor; Vacuolar degeneration
- Death: 8 months
- Progressive motor neuronopathy (PMN)
28
- CNTF knockout: Mouse chromosome 19
- SOD transgenic
- Neurofilament heavy chain
: Overexpression
- PQBP-1 transgenic
35
- PQBP-1
- Transcription and/or mRNA processing factor
- Interacts with polyglutamine disease gene products
- Clinical: Progressive weakness with increasing age
- Pathology: Neuronal loss in spinal anterior horn; Loss of motor axons
- PQBP-1
Patient information: Spinal muscular atrophy
Return to Neuromuscular Home Page
Return to Motor Syndromes
Return to Polyneuropathy Index
References
1. Neurology 1999;53:2187-2189
2. Human Mutation 2000;15:228-237, J Med Genet 2003;40:e39-e42
3. Neurology 2000;54:1534-1537
4. Am J Med Genet 2000;92:117-121; Am J Hum Genet 2010; Online March
5. Am J Med Genet 1998;75:193-195
6. Brain 2000;123:1612-1623, Nature Genetics March 2004
7. JNNP 1998;64:217-220
8. Human Mutation 2000;16:253-263
9. JAMA 2000;284:1664-1669
10. Acta Neuropathol 2000;100:603-607
11. Brain 2000;123:2160-2170
12. Brain 1992;115:1889-1900
13. Pediatr Neurol 2001;24:371-372
14. Pediatr Neurol 2001;25:59-61
15. Neuromuscular Disorders 1998;8:405-408
16. Nature Genetics 2001;29:160-165, Nature Genetics 2001;29:166-173
17. Am J Hum Genet 2002;70:January, Exp Neurol 2014;262 Pt B:91-101
18. J Neurosci 2001;21:9246-9254
19. Neuromuscular Disorders 2002;12:26-30
20. J Neurol 2002;249:290-293
21. Ann Neurol 2002;51;585-592
22. Brain 2002;125;1320-1325
23. Hum Mol Genet 2002;11;1605-1614
24. J Cell Biol 2001;152:1107-1114
25. American Journal of Medical Genetics 2002;110:301�307, Neurology 2022 Jul 14
26. Brain 2002;125:1624-1634
27. J Neurochem 2002;82:1229-1238, J Neurol Sci 2006; Online January
28. Nature Genet 2002;On-Line October 21
29. Hum Mutation 2002;On-Line #552
30. J Mol Biol 2002;324:247�256
31. Ann Neurol 2002;52:680-683
32. Arch Neurol 2002;59:1921-1926
33. J Neurological Sci 2003;209:13-17
34. Nature Genet 2003;On-Line March 10
35. Hum Molec Genet 2003;12:711�725
36. Am J Hum Genet 2003; Online June; Science 2009:323; 1205-1208 & 1208-1211, Neurology 2010; July 28, Neurobiol Aging 2015 Aug 15
37. Nature Genet 2003; Online June 29
38. Am J Hum Genet 2003; Online July
39. Muscle Nerve 2004;29:323�328, Neuromuscul Disord 2022;33:148-152, Mol Genet Metab 2023;138:107525
40. PNAS 2004
41. J Med Genet 2004;41:224�229, Neurology 2009;72:246�252
42. Neuron 2004;41:687-699
43. J Med Genet 2004;41:315�320, Am J Hum Genet 2004; Online September
44. Am J Hum Genet 2004; Online April
45. J Peripher Nerv Syst 2004;9:122-123
46. Hum Mol Genet 2004;13:1677-1692
47. Am J Med Genet 2004; Online Sept
48. J Biol Chem 2004; Online Aug, Neurobiology of Aging 2010; Online Apr
49. Hum Mol Genet 2004;13:R195-R202
50. Nature Genet 2005; Online March
51. NeuroReport 2005;16:657-661
52. Nature Genetics 2005; Online October
53. Neurology 2005;65:1954�1957
54. Brain 2006; On line Feb 22, Neurology 2006; On line January 18, Neurology 2009;72:1669�1676
55. Nature Genetics 2006; Online Feb 26, Ann Neurol 2011;70:964�973
56. J Cell Biol 2006;172:733-745
57. Neurology 2006;67:120�124, Am J Hum Genet 2007; Online May, Eur J Neurol 2020 Nov 21, Ann Clin Transl Neurol 2020 Dec 4
58. Neurology 2006;67 On line June 28 , PLoS ONE 2010;5:29872
59. Hum Genet 2007 Mar 13, Brain 2022 Nov 16
60. Amyotroph Lateral Scler 2007;8:73-78
61. J Neurol Sci 2008 Feb 6
62. Ann Neurol 2008 Online Feb 20, Science 2008 Online Feb 27, Nat Genet 2008 Mar 30, Lancet Neurology 2008 Online Apr 5, Neurology 2012;78:1519�1526
63. Am J Hum Genetics 2009;;84:85-88
64.Neuromuscul Disord 2009 Mar 20
65. Neurology 2009;72:1153�1159
66. Hum Mol Genet 2009;18:1288-300
67. Neuromuscul Disord 2009;19:193-195
68. Am J Human Genet 2009; Online July
69. Am J Human Genet 2009; Online August
70. Brain 2010 Online January
71. Neurology 2010;74:502-506, Cell Rep 2017;20:2100-2115
72. Am J Hum Genet 2010; Online February
73. PNAS 2010; Online April
74. Nature 2010;465:223-226, J Neurol Neurosurg Psychiatry 2011 May 25
75. Neurology 2010;75:539-546, Neurology 2012;78 On-line March
76. J Neurol Neurosurg Psychiatry 2010;81:572-577
77. Neurology 2010;75:611�618
78. Neuron 2010;67:575-587, Neurol Genet 2022;8:e200011
79. Neurology 2011;77:334-340, European Journal of Human Genetics 2012; Online April, Ann Neurol 2014; Online Nov
80. Neurobiology of Aging 2011; Online August
81. Ann Neurol 2011; Online August, J Peripher Nerv Syst 2022 Oct 12
82. Nature 2011; Aug 21
83. Neuron 2011; Online September: A, B, Acta Neuropathol 2012; On-Line Jan, Brain 2012; Online Feb, Am J Human Genet 2013; Online February
84. PLoS One 2011;6(10):e26164
85. J Neurol Neurosurg Psychiatry 2011 Oct 25
86. PNAS 2011; On-line November
87. Annals Neurology 2011; On-line November
88. Hum Mol Genet 2012; Online Mar
89. Nature Genetics 2012; Online Apr
90. Brain 2012;135:1714-1723, American J Hum Genet 2013; Online May A, B, C, Brain 2014; Online Dec, Neurology 2016;87:2235-2243
91. Am J Human Genet 2012; On line June, Neuromuscul Disord 2022;32:806-810
92. PNAS 2009;106:2794�2799
93. Brain 2012; Online June 26, J Med Genet 2012; Online Dec
94. Neurobiol Dis 2012; Online July
95. Nature 2012; Online July, Eur J Neurol 2022 Sep 29
96. Am J Human Genet 2012; Online Aug
97. Am J Human Genet 2012; Online Aug
98. Am J Human Genet 2012; Online Nov
99. Neurogenetics 2012; Online November
100. Mitochondrion 2013 Feb
101. Acta Neuropathol 2013;125:523�533
102. Acta Neuropathol 2013 May 15
103. Am J Human Genet 2013; Online May
104. Neurogenetics 2013 Aug 24, J Neurol 2014 Aug 23
105. Am J Human Genet 2013; Online Oct, Front Neurol 2022;13:865264
106. Am J Human Genet 2013; Online Oct
107. Nat Neurosci 2013 Nov 3
108. Acta Neuropathol 2014 Jan 3
109. Neurobiol Aging 2013 Dec 4
110. Neurology 2014; Online Feb
111. Neurology 2014, Genes (Basel) 2022;13(5)
112. Neurology 2014; Online May
113. Mol Genet Metab 2011;102:6-12
114. Nat Commun 2014;5:4287
115. Neuromuscular Disorders 2014: Online July
116. Neurology 2014;83:990-995
117. Neurology 2014; Online Oct, Eur J Neurol 2022;29:2056-2065
118. Am J Human Genetics 2014; Online October
119. Neuron 2014;84:324-331
120. Science 2015; Online Feb
121. Nature Neuroscience 2013;16:851-855
122. Neurology 2015;84:2430-2437
123. Acta Neuropathol 2015 Sep 10
124. J Clinical Neuromuscular Disease 2015;17:69-71, Neurology 2016; Online June 8, Neurol Genet 2021;7:e599, Neuromuscul Disord 2022;32:527-532
125. Neurology 2015; Online December
126. Am J Human Genet 2016;98:473-489, Eur J Med Genet 2022 Jun 8
127. Parkinsonism Relat Disord 2016 Mar 3
128. PLoS One 2016;11(3):e0151376
129. Am J Hum Genet 2016; Online March, J Hum Genet 2022 Jan 28
130. Neurology 2016 Jun 17, Mol Neurobiol 2023 May 12
131. Amyotroph Lateral Scler Frontotemporal Degener 2016 Jun 27
132. Hum Mol Genet 2016;25:1559-1573, Ann Neurol 2019 Dec 3
133. Am J Hum Genet 2016; Online August
134. Am J Hum Genet 2016; Online September
135. Neuromuscular Disorders 2016: Online September
136. J Hum Genet 2016 Dec 8
137. JAMA Neurol 2017; Online Feb
138. J Neuromuscul Dis 2016;3:487-495
139. Brain 2017;140:1252-1266
140. J Neurol Neurosurg Psychiatry 2017 Apr 1
141. Sci Transl Med 2017;9(388)
142. Sci Rep 2017;7:2116
143. Clin Genet 2018;93:301-309, Ann Neurol 2017 Dec 28
144. Neurol Genet 2017;3(4):e172
145. Neurobiol Aging 2017 Dec 27
146. Neurology 2018 Jan 19
147. J Neurol Neurosurg Psychiatry 2017;88:99-105
148. JIMD Rep 2017 Dec 7
149. Am J Hum Genet 2018 May 3
150. Hum Mol Genet 2016;25:2985-2996
151. Genet Med 2018 Mar 8
152. Neurology 2017;88:1226-1234
153. Neurobiol Aging 2019 Mar 11, Front Neurol 2024:15:1284459
154. Nat Commun 2019;10:1777
155. Brain 2019 Jul 23
156. J Hum Genet 2019 Aug 17
157. Neurology 2019 Oct 2
158. Cell Rep 2019;26:2298-2306
159. Ann Neurol 2020 Jan 19, J Neurol 2020 Sep 30
160. Neuromuscul Disord 2020 Jan 17
161. Neuromuscular Disord 2020; Feb, J Hum Genet 2021 Mar 20
162. Brain 2020 Mar 18
163. Front Neurosci 2020 Apr 28;14:316
164. J Clin Med 2020;9:E2222
165. Amyotroph Lateral Scler Frontotemporal Degener 2020;30;1-8, Genes (Basel) 2021;12:1935
166. Neuron 2020 Nov 25:S0896-6273(20)30883-7, Neuron 2021;109:1945-1946
167. Eur J Neurol 2020 Dec 28
168. Int J Biochem Cell Biol 2020 Jun;123:105746
169. Orphanet J Rare Dis 2021;16(1):10
170. Brain 2021 Jan 18, Brain 2021 Jan 18
171. Front Cell Dev Biol 2020 Sep 9;8:571004
172. J Mol Neurosci 2021 Jan 22
173. Orphanet J Rare Dis 2021 May 7;16:207
174. Ann Neurol 2021 May 28, Ann Clin Transl Neurol 2022 Sep 22
175. Neurol Genet 2021;7:e598
176. Neurobiol Aging 2021 Jun 1
177. Sci Rep 2021;11:13613, Sci Rep 2021;11:11868
178. Semin Pediatr Neurol 2021;37:100878
179. J Neurol Neurosurg Psychiatry 2021 Sep 13, Front Genet 2023;14:1208673
180. J Biol Chem 2021;101299
181. Front Genet 2021;12:746060
182. J Child Neurol 2020;35:717-723
183. Sci Rep 2020;10:20738
184. N Engl J Med 2020;383(2):109-119
185. Am J Hum Genet 2022 Jan 27, Mol Genet Genomic Med 2023;11:e2131
186. NPJ Genom Med 2022;7:8
187. Hum Mutat 2022 Jul 11
188. J Med Genet 2022 Jul 25
189. Front Neurol 2022 Aug 3;13:939775
190. Sci Rep 2022;12:14739
191. Genet Med 2022 Nov 18
192. Brain 2023 Feb 17
193. Acta Neuropathol 2023 Mar 31, Brain 2023 Dec 11
194. Cells 2023;12:847
195. Hum Mol Genet 2023 Apr 18
196. Molecules 2023;28:5801; Rev Neurosci 2023 Aug 2
197. Neurol Sci 2023 Sep 20
198. Front Neurol 2023;14:1242472
199. RMD Open 2023;9:e003431
200. J Med Genet 2023 Oct 27
201. Mov Disord 2022;37:384-391, Ann Med Surg (Lond) 2022;84:104840
202. Neuromuscul Disord 2024:34:114-122
3/26/2024